
II. Ore systems and processes
(Leader Academician A.A.Marakushev)Marakushev A.A. Petrologic formation models of gigantic ore deposits.
A formation model has been developed for gigantic copper-nickel-platinum-bearing deposits of the Norilsk type resultant from fluid sulfurization of iron-rich ultrabasic magmatic differentiates. The model is based on D.S. Korzhinsky's hypothesis on transmagmatic fluids and metamagmatism.
The model takes into account the results of trap formation boring (Norilsk region) revealing volcanic analogs of the Norilsk ore-bearing intrusions which demonstrate an increased content of a number of ore metals (platinum, palladium, chromium). The established trend reflects the magmatic ore concentration process proper and corresponds to sulfide free and sulfide-depleted ore horizons on the Norilsk intrusions and Bushveld. In Bushveld this trend correlates with iron-rich platinum-bearing differentiates (hortonolitic dunites). In the Norilsk sulfide ores the revealed trend of the magmatic ore concentration combines with a gigantic concentration of copper and chalcophilic metals in these ores, inexplicable in the framework of the magmatic processes themselves. Such a complex polygenic character of the Norilsk sulfide ores metallic composition can be explained only with allowance for transmagmatic surfurization of ore-bearing iron-rich magmatic differentiates corresponding to platinum-bearing hortonolitic dunites.
Such sulfurization was directly supported by our findings of relict chromites compositionally close to spinel-chromite mineralization of hortonolitic dunites. So, the developed sulfurization model is original and important for the solution of the general problem of genesis of rich sulfide ores always containing gigantic amounts of iron which can only be attributed to its magmatic concentration. In this aspect, however, the model needs to be further detailed in special experimental studies.
Bushveld, South America, is one of the complex differentiated intrusions wherein the evolution of platinum mineralization is correlated with the evolution of magmatic ultrabasic extremely iron-rich differentiates (hortonolitic dunites, etc).
Fig.1. Petrochemical diagrams (in at%) of the Talnakh (left) and Bushveld (right) ore-bearing intrusions. The crystallization differentiation (shown arrows) is completed by layering of residual melts (shown by solid line) with isolation of extremely iron-rich differentiates also enriched in platinum metals. In the Bushveld massif platinum-bearing hortonolitic dunites formed on their base, whereas in the Talnakh intrusion the melt suffered sulfurization with the formation of platinum-bearing copper-nickel sulfide ores.
The concentrations of platinum metals in the Bushveld intrusion reflect their multistage accumulation both in the course of primary basite-hyperbasite layering of the magmatic chamber in the mantle (with isolation of dolerite-picrite metals which selectively extract sulfur) and at subsequent massif layering with isolation of ore-concentrating sulfide-bearing horisons (sulfide-depleted chromitic and pyroxenitic ones) and extremely iron rich (hortonolitic) dunites. The Bushveld intrusion is unique in having such ferruginous rocks as hortonolitic dunites formed in it. This is due to its isolation from ascending transmagmatic fluid fluxes under the action of which extremely iron-rich melts, corresponding to them, must lose stability and be subject to sulfurization. Such interaction may result in the formation of complex (heterogeneous) ores whose metallic composition is due to two processes, i.e., magmatic differentiation of the Bushveld type and metals introduction by transmagmatic fluids in connection with sulfurization of iron-rich melts. The combination of these processes may endow the mineralization with an extremely large scope leading to the formation of gigantic deposits, for example, the copper-nickel Talnakh deposit in the Norilsk region [1].
The Talnakh intrusion, like the Bushveld, is co-ordinated with a gigantic surface-isometric platform structure deflection filled with thick sedimentary-volcanogenic strata of trap formation. This is typical for such intrusions having preceded in their evolution the mass blanket basalts outflow.
The comparison of the petrochemical diagrams of the massifs is given in fig.1. One can see their general similarity and, also, that the position of the Bushveld hortonolitic dunites (in the region of ferruginous compositions) on the diagram of the Talnakh intrusion is occupied by copper-nickel sulfide ores. Their correspondence is seen not only from the general metallic composition but, also, from the mineralogic specificity in platinum mineralization that differs them from other ore types (chromites and clinopyroxenites).
32
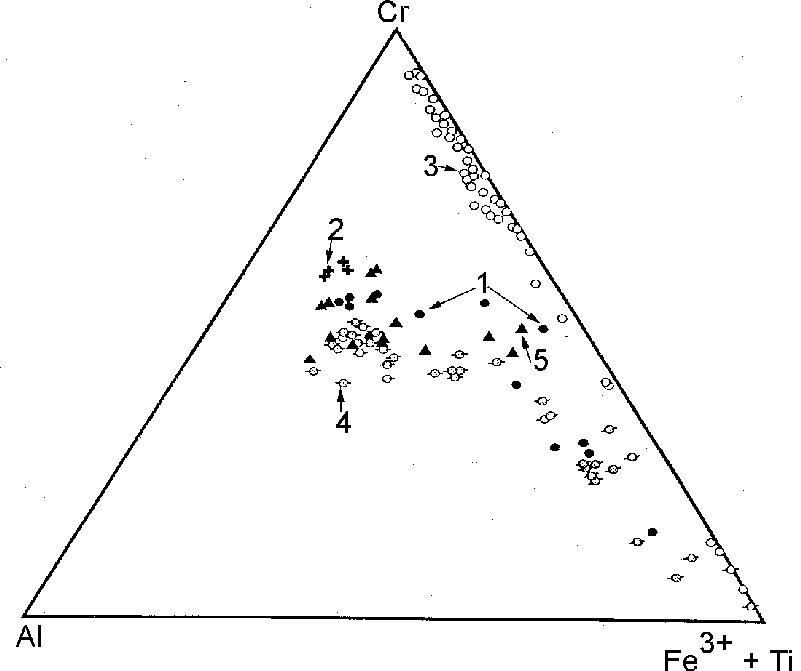
Fig.2. Compositional variations of chrome spinel in hortonolitic dunites (1) and their chromite subordinates (2) from the East Bushveld pipes, in the sulfide ores of komatiites in Australia (3), and sulfide-bearing, platinum-bearing picrites (4), and chromite segregations in them (5) in the Norilsk province (intrusions: Norilsk-1, Talnakh, Manturov).
Also illustrative are compositional variations of chrome spinel represented in hortonolitic dunites (and olivinites) by chrome spinel-magnetite solid solutions series which is close to chromite-magnetite series of these minerals inherent in sulfide ores (fig.2). This correspondence in specificity of platinum metals and chrome spinel unambiguously shows the genetic essence of the sulfide Norilsk ores, reflecting their connection with sulfurization of ferruginous melts-differentiates of picrite-gabbrodolerite sill, rich in platinum metals and iron, under the action of transmagmatic fluid fluxes. In normative minerals symbols this can be expressed by the following reaction: FeMgSiO4+H2S=FeS+MgSiO3+H2O.
Accordingly to the reaction, the initial ultrabasic melts (ferrotroctolite, ferropicrite, ferrodunite), first compositionally homogeneous but iron-rich and therefore, possessing high chemical affinity to sulfur, are subjected to sulfurization and lose the properties of ideal solutions. In the sulfurization dynamics under the action of transmagmatic fluid fluxes they develop liquid immiscibility which is fixed they develop liquid immiscibility which is fixed by textures of droplike impregnate ores with a simultaneous descent of sulfide droplets and the formation in the sill bottom of solid sulfide ores which could, also, form independently due to sulfurization of differentiates, particularly iron-rich ones. Melt sulfurization is accompanied by introduction of copper and chalcophilic metals with simultaneous evolution of effective extraction by sulfide melts of nickel and platinum melts from silicate melts wherefrom they separate due to liquid immiscibility.
The scheme of the development in metals of sulfide-silicate liquid immiscibility in the dynamics of their sulfurization enables one to avoid the ideas of unreally high concentration of ore components (metals and sulfur) in silicate melts, parental with respect to gigantic deposits of copper-nickel sulfide ores. Gabbrodolerites from the Norilsk deposits containing abundant sulfide droplets formed as a result of sulfurization of ferropicrite melts simultaneously with ore differentiations contained in them. Therefore, the scales of the sulfide mineralization in said-type massifs are dictated only by the amount of iron accumulated in the course of differentiation of ferrotroctolite magmas, accordingly, the volume of mineralization can be unlimitedly large.
The ores from the Talnakh deposit are complex, they contain metals, concentrated in the course of the magmatic melt differentiation, as well as metals, introduced together with sulfur by transmagmatic fluid fluxes.
Reference:
#Persikov E.S., Bukhtiyarov P.G. Effect of water and hydrogen on rheologic and other physicochemical properties of magmatic melts.
We have carried out original experimental studies of the temperature (in an albite - NaAlSi3O8 melt) and concentration (in a complete series of basicity of magmatic melts from acidic to ultrabasic compositions -Ab - Ab80Di20 -Di - picrite) dependences of hydrogen solubility at PH2 to 5 kbar and T=1125-1400oC. We have shown that solubility of H2 in an albite melt grows with the temperature which is the converse of the temperature dependence of solubility of H2O (fig.1). We have also experimentally established for the first time that solubility of H2 under isothermal conditions grows as the basicity of magmatic melts grows which is the converse of the concentration dependence of solubility of H2O. (fig.2). In fig.2. the composition of melts and their structural features are reflected by of the well-known structure-chemical parameter - 100 NBO/T (degree of depolymerization) and the hydroxyl OH-reflects the chemically dissolved water.
We have first studied experimentally the effect of PH2O and PH2 on viscosity of Ab80Di20 melts at Pfl to 4 kbar and T=1200-1400oC. The viscosity of the melt in question is shown to decrease appreciably with growing PH2O, i.e. by 1.5 order of magnitude at PH2O=4 kbar and T=1300oC as compared with its viscosity under normal pressure. A small (by approximately 1.5 times) decrease of the Ab80Di20 melt viscosity is also established under a hydrogen pressure (PH2=4 kbar, T=1400oC) which is comparable with the effect of lithostatic pressure on the viscosity of this melt [1].
#The work has been supported by the RFBR (project N 97-05-64448).
33
A theoretical analysis of these and the earlier data on solubility of water and hydrogen in model and magmatic melts of various compositions have made it possible to establish new extremely important features of the mechanisms of dissolved water and hydrogen in them. In particular, we have proved the manifestation of the amphoteric nature of H2O at its dissolution in magmatic melts in the comlete series of their basicity from acidic to ultrabasic compositions. Water is the base with respect to melts in the series: acidic-basic compositions, and its dissolution leads to an increase of their depolymerization degree (basicity growth) and, therefore, to a decrease of activation energy and viscosity. In contrast to it water, dissolving in ultrabasic melts, is the acid with respect to them, and its dissolution is accompanied by a decrease of their basicity and the corresponding growth of viscosity and activation energy. Hydrogen dissolves in melts in significantly smaller degree as compared to water (by approximately two orders of magnitude) and, depending on the melt compostion, different mechanisms of its dissolution will take palce: physical disoolution will dominate in melts free of elements of variable valence (Fe, Ni, Co), chemical dissolution in the form of hydroxyl OH- will dominate in melts with elements of variable valence [2,3].
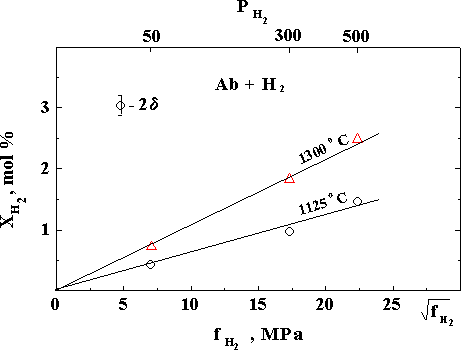
Fig. 1. Hydrogen solubility in an albite melt. (XH2 - molar amount of H2 in the melt, fH2 - fugacity of H2 in gaseous phase, MPa; PH2 - MPa).
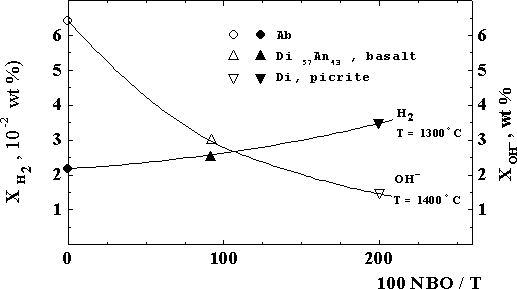
Fig. 2. Structure-chemical dependences of the H2O (as hydroxyl OH-) and H2 solubilities for model and magmatic melts (explanation in the text).
References:
#Persikov E.S., Bukhtiyarov P.G. Rheology and structure of mantle and earth crust magmas: a viscosity study of Ab-Di melts under ultrahigh pressures.
Temperature and pressure dependences of viscosity of melts of middle composition (Ab80Di20) have been studied experimentally for the first time at P to 50 kbar and T=1600-1850oC. It has been found that the temperature dependence of the melt viscosity has an exponential behaviour, the preexponential factor value being constant. The values are obtained for activation energies at pressures 30 and 50 kbar. With account taken of the earlier results on viscosity of Ab-Di melts of other compositions, it has been shown that minimum viscosity of the melt in question will be at P=60 kbar (fig.1.). The viscosity of the melt Ab80Di20 decreases appreciably with the growing pressure. For example, at P=50 kbar and T=1800oC the viscosity of such melt decreases by 1.5 order of magnitude as compared with its viscosity under normal pressure.

Fig.1. Isothermal (1850��) dependence of the viscosity of Ab - Di and Jd melts on the pressure (open symbols - extrapolated values, see in the text for explanation).
#The work has been supported by the RFBR (project N 97-05-64448).
34
The data obtained on the dependence of viscosity of model melts of the albite-diopside system and also, jadeite melts on lithostatic pressure are illustrated in fig.1. They show that viscosity decreases significantly with the growing lithostatic pressure throughout the range of compositions with the exclusion of diopside melt for which the pressure dependences of the viscosity exhibit the inverse behaviour, i.e. as the lithostatic pressure grows, the viscosity values of diopside melt increase. It should be mentioned that the inversion of the pressure dependence of viscosity of Ab-Di melts was first experimentally proved at pressures to 15 kbar in the work [1], and for a jadeite melt this dependence was predicted in the work [2] and experimentally proved by the same authors in the University of Tokyo (fig.1). The principally new result of this work is that the inversion of the pressure dependence is proved also for the activation energy of the viscous flow of these melts. An analysis of the diagram (fig.1) shows that pressure magnitude in viscosity minima points is strongly dependent on the melt composition, decreasing with the growth of its basicity. An increase with the pressure of the viscosity of ultrabasic melts and, also, of more polymerized melts after the minima is in full agreement with theoretical prerequisites whereas a considerable decrease of viscosity of alumosilicate and magmatic melts with the growing pressure is anomalous at the first stage. The nature of this anomaly is a debated topic. A most possible mechanism capable of explaining this anomaly is a structural transition in four-coordinated aluminium in the melts to six-coordination with respect to oxygen, i.e. the transition of Al from the position of network forming cation to that of cation-modifier that, naturally, is accompanied by an increase of the melt depolymerization degree. One may, however, agree with opponents of this idea [4] in that the structural transitions AlIV Al VI and SiIVSi VI are not a comprehensive explanation of the nature of the anomaly of the pressure dependence of viscosity for all model alumosilicate and magmatic melts. A theoretical analysis of the results of this work has shown that the structural AlIVAl VI transition fully explains the anomaly of viscosity for melts with molar aluminium-to-silicon ratio
Al VI and SiIVSi VI are not a comprehensive explanation of the nature of the anomaly of the pressure dependence of viscosity for all model alumosilicate and magmatic melts. A theoretical analysis of the results of this work has shown that the structural AlIVAl VI transition fully explains the anomaly of viscosity for melts with molar aluminium-to-silicon ratio 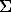 AlIV/(AlIV+SiVI) >0.33, in particular, for jadeite melts. With e<0.33 there may hold the idea, proposed in [4], weakening of O-Al-O, O-Si-O bonds due to diminution of their angles and melt structure ordering with the growing pressure. The calculations show that a relative role of the second mechanism has to diminish with a growth of melts basicity. So, for example, for an albite melt at P=95 kbar, which corresponds to minimal values of viscosity and activation energy, the complete AlIVAlVI transition is responsible for about 70% of the observable viscosity decrease whereas for a Ab30Di70 melt the complete AlIVAlVI transition at Pmin=15 kbar is responsible for already about 90% of the experimentally established viscosity decrease. Undoubtedly, direct structural studies of alumosilicate and magmatic melts at high T and P in "in situ" runs are required for an incontroversial understanding of the mechanism of the anomalous dependence of viscosity of such melts on pressure.
AlIV/(AlIV+SiVI) >0.33, in particular, for jadeite melts. With e<0.33 there may hold the idea, proposed in [4], weakening of O-Al-O, O-Si-O bonds due to diminution of their angles and melt structure ordering with the growing pressure. The calculations show that a relative role of the second mechanism has to diminish with a growth of melts basicity. So, for example, for an albite melt at P=95 kbar, which corresponds to minimal values of viscosity and activation energy, the complete AlIVAlVI transition is responsible for about 70% of the observable viscosity decrease whereas for a Ab30Di70 melt the complete AlIVAlVI transition at Pmin=15 kbar is responsible for already about 90% of the experimentally established viscosity decrease. Undoubtedly, direct structural studies of alumosilicate and magmatic melts at high T and P in "in situ" runs are required for an incontroversial understanding of the mechanism of the anomalous dependence of viscosity of such melts on pressure.

Fig.2 Generalized (prediction) diagram of the dependence of the viscosity of near-liquidus acidic (jadeite-Jd) and ultrabasic (diopside-Di) melts on pressure in the entire range of erath crust and upper mantle depths (see in the text for explanations)
We have carried out a theoretical analysis of the pressure dependence of the viscosity of magmatic melts in the whole range of their basicity from acidic to ultrabasic compositions and developed foundations of a new structural-chemical model for calculation and prediction of magmas viscosity throughout the entire range of earth cryst and upper mantle depths. Using the proposed model, a generalized (prediction) dependence of the viscosity of near-liquidus (by 50oC above the corresponding melting temperatures) magmatic melts on the pressure has been obtained for the entire range of earth crust and upper mantle depths, fig.2. An analysis of this diagram suggests that throughout the earth crust and upper mantle depths the viscosity of near-liquidus acidic melts will decrease dramatically (by greater than 8 orders of magnitude) whereas the viscosity of ultrabasic melts will decrease to a much smaller degree (by 2 orders of magnitude). In a more real depths range (to 300 km), where magmas can occur, a decrease of the viscosity of acidic melts will remain quite significant (about 6 orders of magnitude) whereas the viscosity of ultrabasic melts will slightly increase (less than 0.5 order of magnitude) [3].
References:
35
#Stolyarova T.A. Deficiency of volatile components in the composition of apatite.
In our earlier studies of fluorapatites, conducted under the project, we substantiated a new interpretation of isomorphism of fluorapatites as solid solutions with phosphates compositionally close to whitlokite. This interpretation explained the usual deficiency of volatiles in the fluorapatite composition and was taken as the basis in the studies of the year under review, devoted to thermochemistry of chlorapatites. We generalized the data on natural chlorapatites and ascertained that like in natural fluorapatites in chlorapatites a theoretical content of Ca2P3O12Cl (6.8 wt%) is never, practically, achieved. The compositions of even most chlorine-rich apatites, inherent in meteorites, are indicative of incorporation into their composition of calcium phosphates which contain no volatiles and, by analogy with fluorapatites, the chlorapatite formulas may be represented as nAp(1-n)Wh. In the earth's chlorapatites the share of the Wh component is still higher.
In accordance with the generalized natural data which bear witness to the existence of isomorphous chlorapatite-whitlokite series we made up research program for the year 1998 which included synthesis of chlorapatite, whitlokite, and apatites of intermediate composition. Melt and hydrothermal synthesis techniques were used therewith.
The idea of the phosphates incorporation into isomorphous apatite series compositionally similar to whitlokite was taken as the basis at crystallochemical recalculations of chemical analyses of apatites and processing of their thermochemical data.
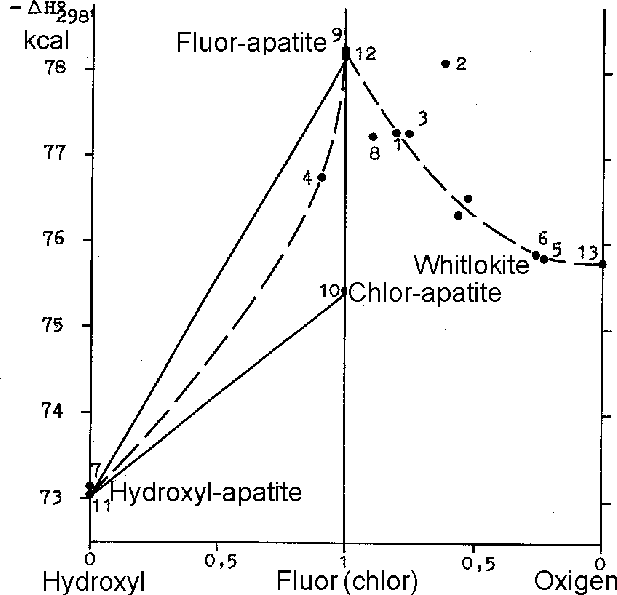
Fig. 1. Change in the standard formation enthalpy (in kcal/at) of apatites from isomorphous series hydroxylapatite-chlorapatite, hydroxylapatite-fluorapatite, fluorapatite-whitlokite.
Determinations of dissolution enthalpy of natural and synthetic apatites were performed on a differential automated microcalorimeter DAK-1-1A fabricated in the experimental plant for scientific instrumentation RAS, Chernogolovka, and reequipped by us for work in aggressive media. Samples dissolution conditions were selected in preliminary runs: concentration of hydrochloric acid water solution - 20 wt%, solvent volume 3.5 ml, charge - 1-3 mg, dissolution temperature - 40oC. Weighing was performed on a "Sartorius" balance with an accuracy of 10-6. The cells were calibrated electrically, the calibration was repeated before and after each measurement.
The diagram of fig.1. gives the comparison of the obtained results including those on apatite solid solutions: hydroxylapatite-fluor(chlor)apatite, and fluor(chlor)-apatite-whitlokite. The latter substitution is energetically similar with hydroxyl group - for- fluorine substitution.
An investigation of the isomophous fluorapatite-whitlokite series unveils the potential of thermodynamic calculation of their real stability at a significant deficit in the composition of fluorapatites.
Thermochemical substantiation of the new interpretation of isomorphism of apatite minerals suggests, also, a broader consideration of apatites as intermediate solid solutions in a more general isomorphous system Ca9P6O24-Ca (F,Cl,OH)2 in which the theoretical composition of apatite results from the reaction Ca9P6O24+ Ca(F,Cl,OH)2 =Ca10P6O24(F,Cl,OH)2 that is accomplished in this form at grows, still smaller amount of Ca(F,Cl,OH)2 gets involved into the reaction, and there arises the considered series of solid solutions nAp(1-n)Wh compositionally approaching whitlokite. The effect of temperature can be judged upon by a decrease of the role of volatiles (F2,Cl2,H2O) in their composition. Inasmuch as crystallochemical formulas of apatite and whitlokite contain the same atomic quantity of phosphorus, it should be taken as the basis in calculating the summarised formulas of apatite-whitlokite solid solutions.
##Konnikov E.G. and Pal'yanova G.A. Melting temperatures of the fluid-pyrrhotite mixtures
Russian Academy of Sciences, Institute of Experimental Mineralogy, Russian Federation
Siberian Board of Russian Academy of Sciences, United Institute of Geology, Geophysic and Mineralogy, Russian Federation
This paper presents the results of the experiments focused on the researches of volatile compounds (H2O, CO2, Cl) influence upon sulphides solidus temperatures. They are interesting for a comprehension of thermodynamic parameters of the natural sulfide melts. Contrary to the traditional view it has been ascertained there is not full water solubility into sulfide liquid. The latter doesn't exceed maxima 10 wt.% of H2O and 15-20 wt.% of carbonic acid, each of them decreases not more than 80-130oC of the pyrrhotite (Po) solidus temperature. The experiments and thermodynamic simulations showed sulfide solidus declining must be accounted for a partial oxidation of sulfide melt iron that results in the pyrrhotite+magnetite (Mt) eutectic appearence. In spite of the previous investigations have not been acquired any evidences of pyrrhotite melting temperature dependence on chlorine.
#The work has been sponsored by the RFBR (project N 97-05-64159).
##The authors would like to thank th Russian Foundation for Basic Research (RFBR) for the financial support (grant N 96-05-64714) their investigations.
36
Introduction. As it has been shown by study of the magmatic sulfide deposits related to layered mafic-ultramafic complexes, formation of the sulfide lodes therein occurred under high volatile components pressure. It is evidenced by abundance of OH- and Cl-bearing silicates at the ambient rocks near sulfide bodies. In the generally accepted opinion the sulfide bodies of the magmatic deposits are resulted by sulfide melt crystallization at very high temperature up to 850-1050oC (Kullerud, 1964; Naldrett, 1969). But thermodynamic condition of some sulfide deposits to be estimated basing on different methods show rather low temperatures which vary from 400 to 750oC (Naldrett, Richardson, 1967). Taking into accout the significant volatile influence on melting temperature of silicate systems, there is a tendency to attribute the low temperature crystallization of natural sulfide liquids at the fluid component expense.
Meanwhile, it is surprising that investigations of the fluid dependence on melting processes in sulfide systems occur rarely. First of all this problem was regarded by F.G.Smith (1963) who had calculated pyrrhotite solidus curve basing on the Schreder equation. According to those calculations, pyrrhotite melting along with 10 wt.% of H2O decreases its solidus temperature by 300oC. But this conclusion has been experimentally refuted by A. J.Naldrett & S.Richardson (1967). These authors have shown that ... water has little influence on melting temperatures of pyrrhotite-magnetite mixtures and that it cannot be appealed to as a flux to explain the occurrence of natural, low-temperature (<900oC) sulfide-oxide magmas(p.431).
Due to a contradiction of the above mentioned author's conclusions, it has got obvious that the researches on this topic must be continued.
Methods. A dimension of fluid solubility into sulfide liquid could be acquired on the basis of character of its solidus temperature change because of sulfide's melt glassing impossibility. All 72 runs on studying of fluid (H2O, CO2, Cl) influence on the melting temperature of sulfides were carried out with nearstechiometric composition (Fe0,92-0,94S) of pyrrhotite synthetized artifically. To compare experimental results the runs are conducted at identical thermodynamic parameters, namely P=1000 atm, T=1000-1160oC, and fO2 was monitored by the NNO buffer. A high-gas-pressure vessel with the interal heating was used for the runs. High melting temperature of the monosulfide urges to use platinum tubes for the charge. In order to prevent pyrrhotite interaction with platinum tube, the charge was isolated by a silica crucible from tube's wall. The fluid phase concentration in the runs varied from 5 to 40 wt.% of pyrrhotite powder weight. The runs duration consists of usually half or an hour and it was limited by the silica crucible stability because silica glass recrystallizes into cristobalite if one undergoes with long heating. But equillibrium in the fluid-sulfide system during that time could be achieved (Naldrett&Richardson, 1967).
Our experiments were conducted according to the 'doubled capsule' method. The fluid-pyrrhotite blends were put into a little (of 5 mm in diameter) capsule and that was welded. Then this capsule was placed inside some wider (of 7 mm in diameter) one along with metallic Ni powder and water and large capsule was welded as well. A start of pyrrhotite melting was fixed by sulfide-magnetite intergrowths between pyrrhotite grains identical to those in A.J.Naldrett experiments (1969).
Experimental results. The executed 15 runs have shown that the melting start temperature of sulfide in the Po+H2O system is declined by 80-100oC as compared to the 'dry' solidus of pyrrhotite under water concentration range up to 0-10 wt.%. Further increasing of the sulfide liquid soludus temperature does not occur (Fig.1). Taking that into account, it might be concluded that H2O solubility threshold does not exceed 10 wt.% at thermodynamic parameters of these experiments. Such our conclusion does not coincide with the previous assumptions (Smith, 1963).
The next experiments (16 runs) were aimed at investigation of CO2 on the melting temperature of Fe-monosulfide and were carried out under Po+H2O+CO2 blend pressure. The water-carbon-dioxide fluid mixture was obtained by the thermal decomposition of oxilic acid (H2C2O4 . 2H2O). The experiments showed that the temperature of pyrrhotite solidus decreased some more by 30-40oC as compared to the one in Po+H2O system and a top solubility limit of the fluid in the sulfide liquid increased up to 15-20 wt.%.
Pyrrhotite melting with 0,5N and 2N HCl water solutions does not display noticible chlorine influence on its solidus temperature. The sulfide solidus curve in T vs Cfl-diagram (Fig.1) coincides exactly with the same for the system 'Po- pure H2O'. Almost full immiscibility of pyrrhotite with chlorine was shown by the series of the runs (27) in the system Po-NaCl at T=700oC under atmospheric pressure (Fig.2). These results are in a good agreement with the experimental data at the Po+H2O+HCl system.
Thermodynamic simulations of the experiments and microprobe analyses of the run products enable to ascertain that pyrrhotite melting temperature decreasing in the runs with oxidized gases is accounted for by Mt-minal occurrence in sulfide liquid due to the partial oxidation of the iron (Fig.3).
The computer simulation showed as well that the gases H2, H2S, SO2, S2 are stable along with H2O, CO2, HCl under the runs PT conditions (Fig.4). It could be seen in this picture all gases concentration enhance according to temperature increasing.
Conclusion.
1. For the first time the data on a solubility of natural fluid in sulfide melt has been obtained. It was first experimentally shown that H2O had got though high (near 10 wt.%) but limited solubility in the pyrrhotite liquid.
2. Our experimental result on solubility CO2 in the sulfide melt is first acquired as well as the data about carbon-dioxide influence on pyrrhotite solidus temperature.
3. Low influence of the Cl-combinations on a start of pyrrhotite melting has been shown.
4. In our opinion all aforesaid experimental results are of an essential contribution into the theory of Cu-Ni sulfide deposits.
37
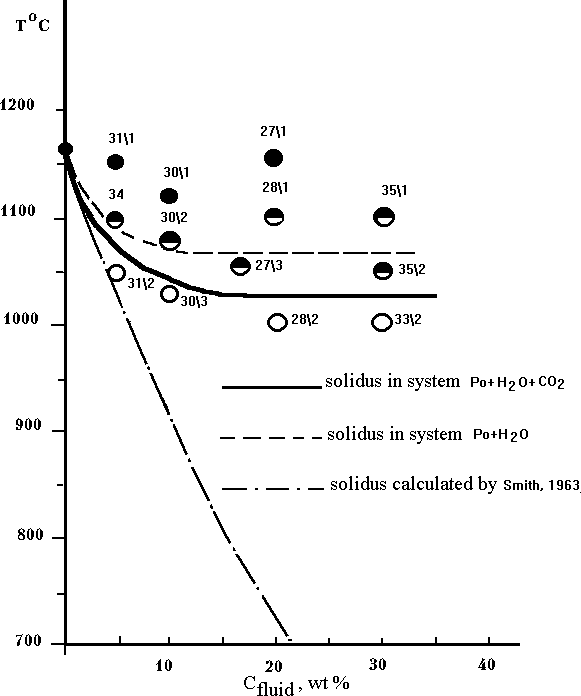
Fig.1. The ToC vs Cfl-diagram for systems Po+H2O and Po+H2O+CO2.
Fig.2. The phase diagramm of Po-NaCl system under atmospheric pressure.
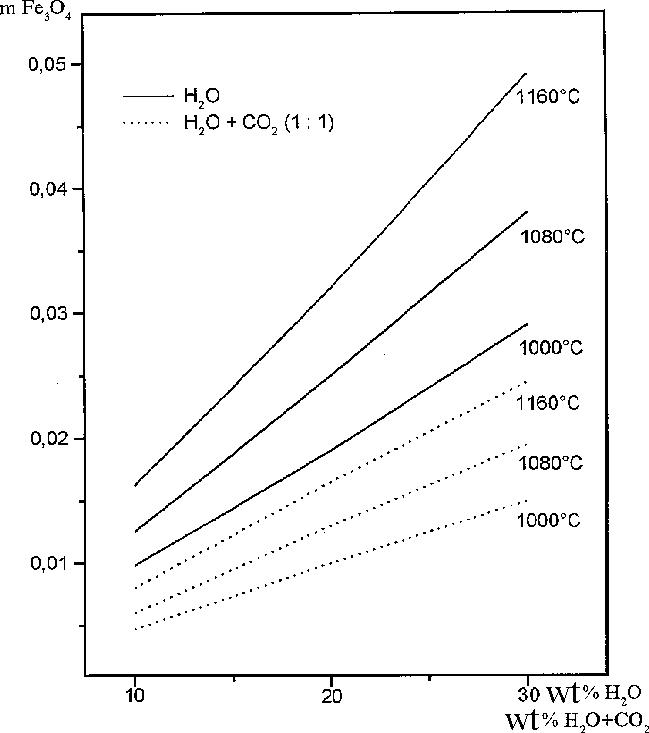
Fig.3. The mole abundance of Mt formed during interaction of Po with H2O and H2O+CO2 at different temperature; the fluid's amount is in wt.%.
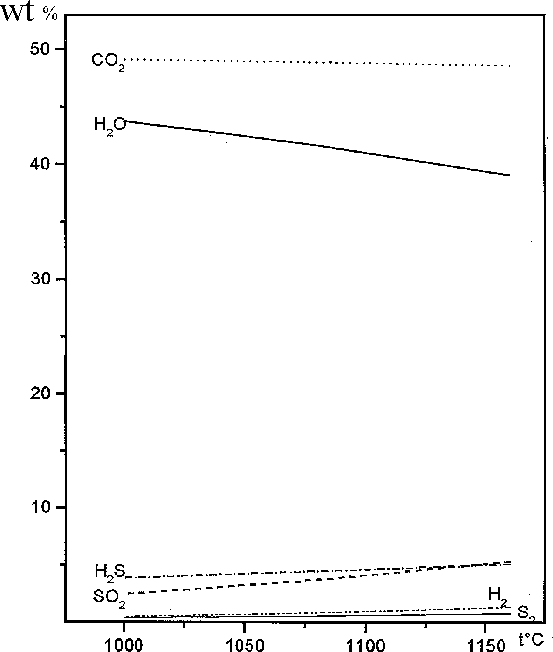
Fig.4. The gas phase composition (wt.%) in system Po+H2O+CO2 depended on temperature.
References:
#Suk N.I. Immiscible liquids in silicate-carbonate systems (experimental study)
Introduction. The possible role of liquid immiscibility in the genesis of carbonatites and associated igneous rocks was discussed by previous authors. Previous experimental evidence confirms that wide immiscible field between silicate and carbonate liquids does exist [6,7,1]. Geochemical review of natural carbonatite complexes indicate accumulation of phases enriched in rare metals (bastnaesite, pyrochlore, baddeleyite and others) in the carbonatites and estimate that some REE or complex REE-Y-Nb ore deposits are connected with intrusives of carbonatite type (for example, complex ring massif Tomtor, Siberia) (Entin et al. 1990). So, investigation of the behaviour of some ore metals (REE, Nb) in immiscible silicate-carbonate systems becomes an important aspect of carbonatite genesis problem.
#This study was supported by the Russian Foundation for Basic Research (project N 97-05-64158).
38
Our experimental study deals with investigation of liquid immiscibility in silicate-carbonate systems at T=1100oC and P=2 kbar and distribution of some ore metals (REE, Nb, Ta) between silicate and carbonate melts. Geochemical data estimate presence of apatite, sulphides, phlogopite in carbonatites, more over, in massif Tomtor there were observed the apatite-carbonate eruptive dykes or tubes and tuff-lavas of phosphate composition [5]. This fact confirms the supplementary fluid components (such as phosphorus and, probably, chlorine and fluorine) enrichment in latter carbonatites. So, the effect of additional fluid components on silicate-carbonate immiscibility and behaviour of ore elements in such immiscible systems are an important problem to be solved. We tried to study an immiscibility in silicate-carbonate systems with addition of phosphate or chloride and distribution of some ore metals (REE, Nb, Ta) between liquid phases. The results obtained are presented in this paper.
Experimental method and results. Silicate-carbonate differentiation of melts was experimentally studied in agpaitic and calciferous systems. The compositions of silicate model melts were determined by mixtures of rock-forming minerals (albite, diopside) and the compositions of carbonate model melts were determined by Na2CO3 or mixtures of Na2CO3 and CaCO3. The distribution of ore metals between immiscible liquids was studied in these systems with addition of La2O3, CeO2, Nb2O5, Ta2O5. Phosphorus and chlorine in some special experiments were added as NaPO3 and NaCl respectively. All starting mixtures were dried for 18-20 h at T=100oC. The experiments were carried out in a high gas-pressure vessel in sealed platinum capsules (d=3 mm) at T=1100oC and P=2 kbar. The weighed portion was 100 mg. Each experiment lasted 6 hs. The products were quenched at an average rate of 200oC/min. The polished samples were analized on a Camebax microprobe equipment with a Link H.E.D.A. system.
A wide immiscibility field was found in studied silicate-carbonate systems, where the starting materials were splitting into silicate and carbonate liquids (Fig.1). The salt melt occured as drops in silicate matrix or formed a large constituents with a distinct phase boundary between both phases. Here the presence of distinct phase boundaries, similar-like compositions of drops and large constituents of each melt, a repetition of the results as well as, obtaining of the similar results by using different initial materials make it possible to conclude about time of experiments enough for endurance of the run to reach equlibrium.
The silicate melt was quenched into homogeneous glass, whereas the carbonate melt formed fine-grained quenched aggregates. Inhomogeneity of the obtained carbonate phases was revealed. This is likely to result from the immiscibility of alkaline and calciferous carbonate melts. The sodium-rich (up to practically pure Na2CO3) and calcium-rich fractions were observed in carbonate phase (Fig.2).
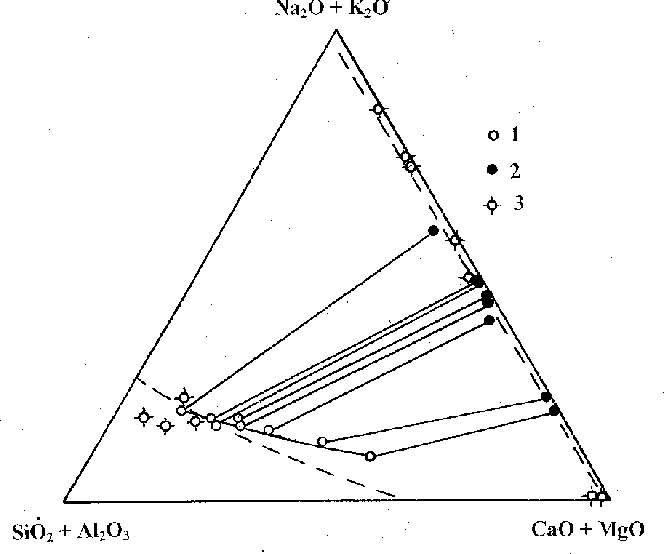
Fig.1. Experimental results on differentiation of melts into (1) silicate and (2) carbonate melts at T=1100oC and P=2 kbar. Tielines connect the compositions of coexisting phases. Dashed line shows the immiscibility field at T=1250oC and P=5 kbar [7]. 3 - compositions of carbonatites and nephelinites of Volcano Oldoinyo Lengai, Tanzania [3,4].
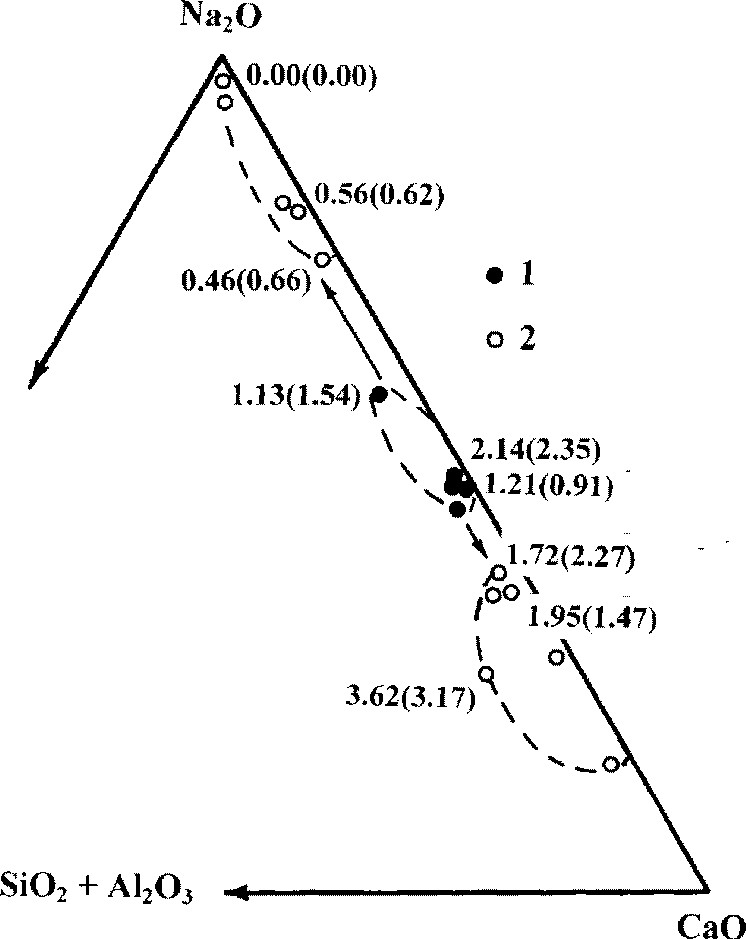
Fig.2. Calk-alkaline separation of carbonate phases (1) into Na-rich and Ca-rich fractions (2) in experiment (arrows). Numbers near points - contents of La2O3 and Ce2O3 (in cramps).
The distribution of REE (La, Ce), Nb, Ta between immiscible silicate and carbonate melts was experimentally studied. It was found that partitioning of REE (La, Ce) between immiscible melts in alkaline systems is in favor of the carbonate melt. The partition coefficients for REE between alkaline-lime carbonate and silicate melts (DREE carbonate melt/silicate melt ) are approximately 1.5-2.5. Our experiments show that REE concentrate in Ca-rich carbonate fractions, while Na-rich fractions are almost La and Ce free (Fig.2). By contrast, in the lime silicate-carbonate systems REE conversely are concentrated in the silicate melts which are also rich in calcium. The results of studing the distribution of Nb and Ta between immiscible phases show that these elements are predominantly accumulated in silicate melts.
39
The study of the effect of phosphorus and chlorine on silicate-carbonate immiscibility and behaviour of ore elements in these systems was based on the results of our experiments in pure silicate-phosphate and silicate-chloride systems [9]. REE, Nb, Ta were found to enrich the phosphate melt in silicate-phosphate systems. Our experimental results illustrate the indifferent behaviour of Nb, Ta and in some degree REE in the chloride extraction in silicate-chloride systems.
In the alkaline silicate-carbonate systems with addition of phosphate the salt phase is inhomogeneous and contains the constituents of carbonate and phosphate compositions. The phosphate phase is more effective in concentration of REE than carbonate one, and enriched in these elements.
In alkaline silicate-carbonate systems with addition of chloride the separation of carbonate-chloride melt from silicate one is observed. Salt liquid is inhomogeneous phase and is divided into chloride (NaCl) and carbonate phases. Salt phase is depleted in ore metals (REE, Nb, Ta), which concentrate in a silicate melt. But it was observed that carbonate phase is reacher in REE as compared with chloride phase.
So, our experimental results illustrate positive role of phosphorus and negative role of chlorine in concentration of ore elements by salt melts in silicate-carbonate systems.
Discussion. A wide immiscibility field was found in the studied silicate-carbonate systems, where starting melts were splitted into carbonate and silicate liquids. The separation of carbonate melts from silicate one at high pressure was experimentally studied in previous works at P=5 kbar [7] and at P=15 kbar [1]. Our data are in a good egreement with them (Fig.1). Comparison of these data allows to conclude that pressure increase and temperature decrease promote the expansion of immiscibility field.
The alkaline-lime immiscibility in carbonate melts revealed by the experiments evidently played a certain part in the formation of carbonatites of intrusive ijolite-urtite complexes where carbonatites are presented by calcite and dolomite types. Alkaline carbonates are supposed to migrate with fluids into surrounding rocks, which have been effected by alkali metasomatism (fenitization). Agpaitic character of initial silicate melts was shown by ordinary assosiation of carbonatites and aegirine- and others agpaitic rocks. In genetic aspect it determines the compulsory participation of alkaline carbonates in primary portions of carbonate melts, separeted from silicate melts, accoding to our experiments. In magmatic evolution calc-alkaline carbonate magmas conserve their original composition only in a volcanic setting. For example, carbonate magmas of Volcano Oldoinyo Lengai (Tanzania) [3,4] have compositions comparable with experimental carbonate melts as well as compositions of silicate melts experimentally obtained in agpaitic systems provide good fit to compositions of nephelinites of considered Volcano (Fig.1). There are observed the extrusive lime carbonatites of Volcano which don`t conserve alkaline composition. Their primary alkaline character may be confirmed by their structure and existence of calcite pseudomorphs by replacement of the alkaline carbonates [3]. Such processes of alkaline-lime transformation of carbonatite magmas supposed to realize in intrusive carbonatites of different genetic types [8].
Partitioning of REE between immiscible phases in studied alkaline systems to carbonate melts make it possible to conclude about the formation of REE-deposits exclusively with respect to alkaline (agpaitic) magmatism. The partition coefficients for REE between alkaline-lime carbonate and silicate melts in experimental systems (D=1.5-2.5) are comparable with those for REE partitioning between carbonate ocelli and host lamprophyre at Callander Bay, Ontario (D=2-3) and between carbonatite and coexisting ijolite (D=2.1) at Seabrook Lake, Ontario [2].
References:
40