V. Experimental and theoretical studies of fluid systems
(Leader Dr. Sci. M.A.Korzhinsky)
Korzhinsky M.A. The solubility of iron chloride in H2O-CO2-NaCl system at supercritical conditions.
The investigations of FeCl2 solubility at supercritical conditions in H2O-CO2-NaCl fluid mixture were carried out at constant FeCl2 fugacity according` to the equilibrium:
Fe3O4 + 2AgCl = FeCl2 + Fe2O3 + 2Ag for which
K=f FeCl2
The experiments executed in the H2O-CO2 system at 600oC and 2000 bars and in pure water at 600oC and at the pressures from 1 to 2000 bars have shown that the equilibrium volume concentration of FeCl2 (C mol/l) is defined by volume concentration of water in the systems. With increasing CH2O in both system from 0.02 to 10 the equilibrium CFeCl2 smoothly grows from 2.10-4 to 5.5.10-3. Hydration number for this range is 0.5. In the range of water concentration from 10 to 32 CFeCl2 harply increases from 5.5.10-3 to 1.2.10-1. Hydration number for this range is 3.
With addition to the system of the background electrolyte NaCl (KCl, CaCl2) at constant T, P the equilibrium concentration of FeCl2 increases. The growth of concentration is due to formation of complexes like nNaCl.FeCl2. For the range of NaCl contents (mol/l) 0 to 0.61 n is 1 and for the range from 0.61 to 2.56 n is 0.5.
In the presence of both CO2 and NaCl in the fluid mixture the tendency of changing equilibrium FeCl2 concentration is similar to that of separated systems: decreases with addition of CO2 and increases with addition of NaCl.
Sretenskaya N.G., Zakirov I.V. An investigation of mechanisms and kinetics of the associate formation in supercritical fluids.
The earlier theoretical and experimental studies have shown [1,2] that the process of associate formation takes place in gases having close- to - critical densities, i.e. molecules combine to two-, three- etc. dimensional clusters, depending on the temperature and pressure. In order to quantitatively estimate the degree of clusterization as a function of the T-P parameters we have designed a technique to study the interdiffusion of two gases. Under the condition where one gas is nonideal and the other ideal the effect of their interaction has to show up in an increase of the total pressure due to collapse of the cluster structures.
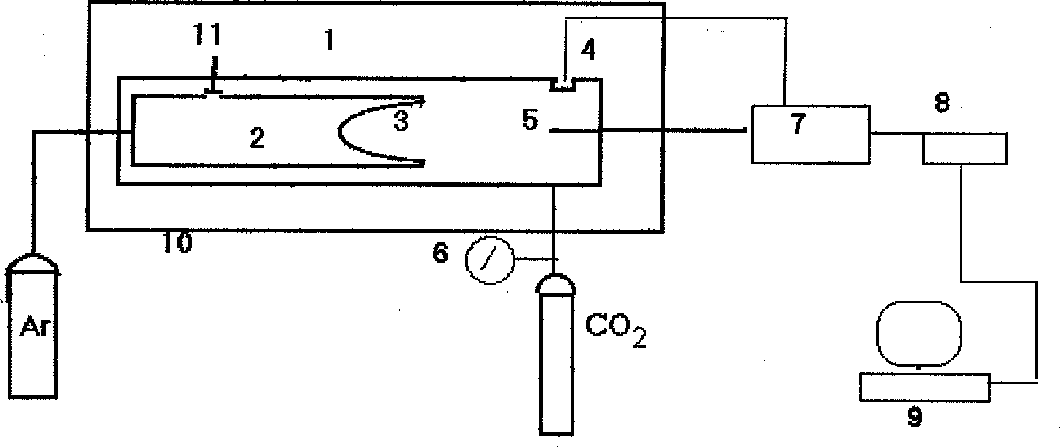
We have constructed an apparatus the schematic presentation of which is shown in fig.1. It consists of reactor 1 with built-in pressure transducer and thermocouple 5. Inside the reactor there is chamber 2 with sensitive membrane 3 and valve 11. The reactor is placed into air thermostat 10. During the run the reactor is filled with CO2 at the appropriate pressure and temperature with the valve 11 closed. Then at the same T-P parameters Ar is fed to chamber 2. The membrane enables the pressure equalization between CO2 and Ar with an accuracy of 0.04 at. Gate 11 is then opened and the gases slowly interdiffuse until a complete mixing ensues. During the run signals from the pressure and temperature transducers are registered every 10 ms by means of an interface consisting of switch 7, analog-digital transducer 8 and an ADT controller built-in into personal computer 9. A software enables one to introduce into the computer memory the whole of the process for two hours, averaging over 10 points each seconds.
The interdiffusion of Ar and CO2 was studied at 25 and 32oC isotherms and initial pressures of 30, 40, 50, 60, and 70 at. The results are a continuous record of the T-P vs time curve. Fig.2 illustrates the results of the run at 25oC and initial pressure 59.3 at. As is seen, the temperature is practically constant in the process of diffusion whereas the pressure grows up to a certain constant value. Analogous results were obtained for all T and P studied. Fig.3. illustrates the dependence of the pressure gain magnitude (in pct) on the initial pressure for two isotherms. The effect of the pressure gain is seen to increase with the initial CO2 density. As at these parameters Ar is ideal and its density is practically constant, the observable effect is attributed to a decrease of the clusterization degree of the dense (nonideal) CO2.
Fig.1. Schematic presentation of the apparatus for a study of interdiffusion of gases. 1-reactor; 2-inner chamber; 3-membrane; 4-pressure transducer; 5-thermocouple; 6-manometer; 7-switch; 8-analog-digital transducer; 9-computer; 10-air thermostat; 11-valve.
So, the unique apparatus enables the present-day automated determination of the quantitative data on the major contribution to nonideality of individual gases and nonideality of homogeneous gas mixtures in the near-critical region.
References:
39
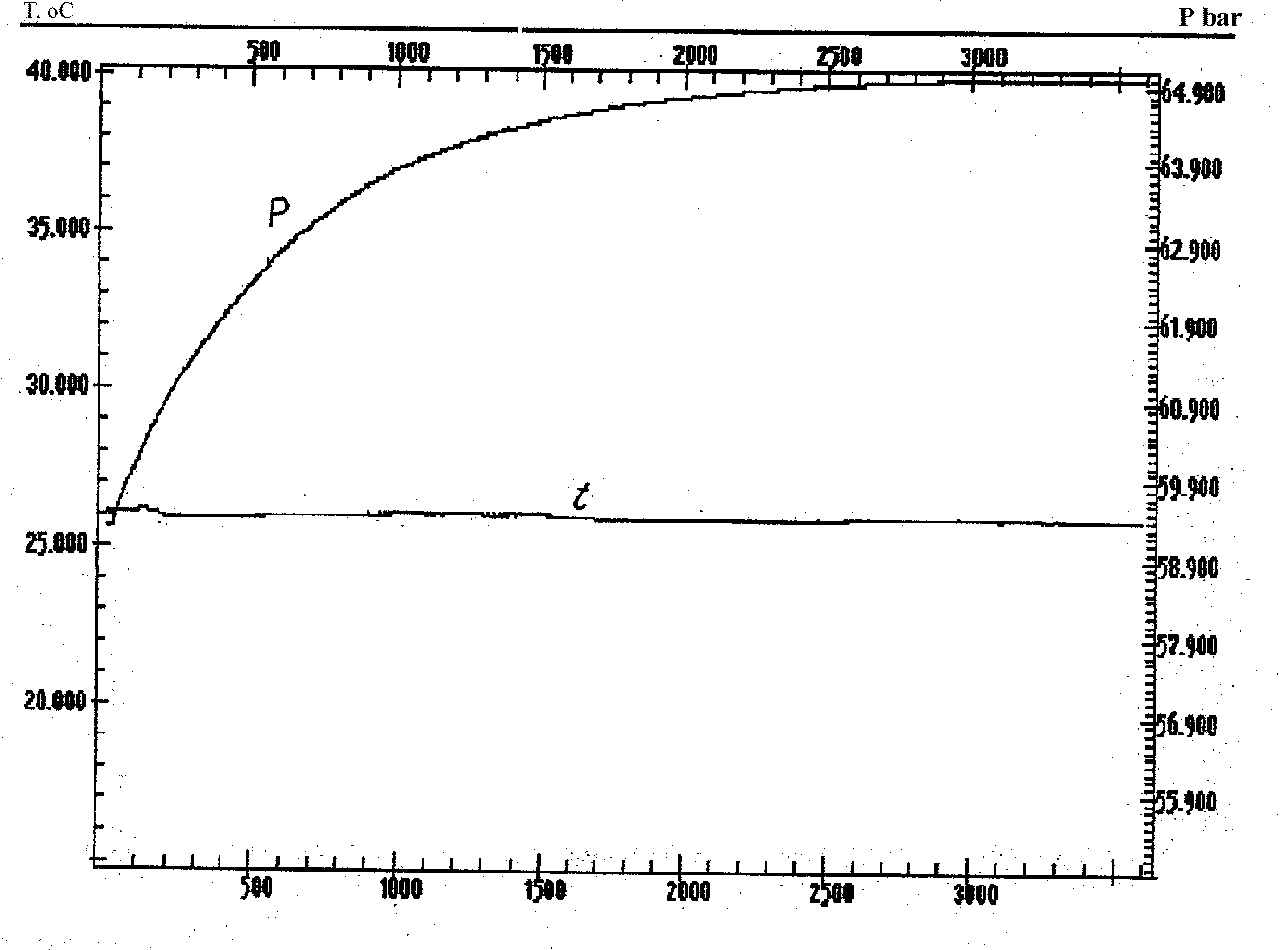
Fig.2. P-T vs time curve in the process of CO2 and Ar interdiffusion at 25 oC.
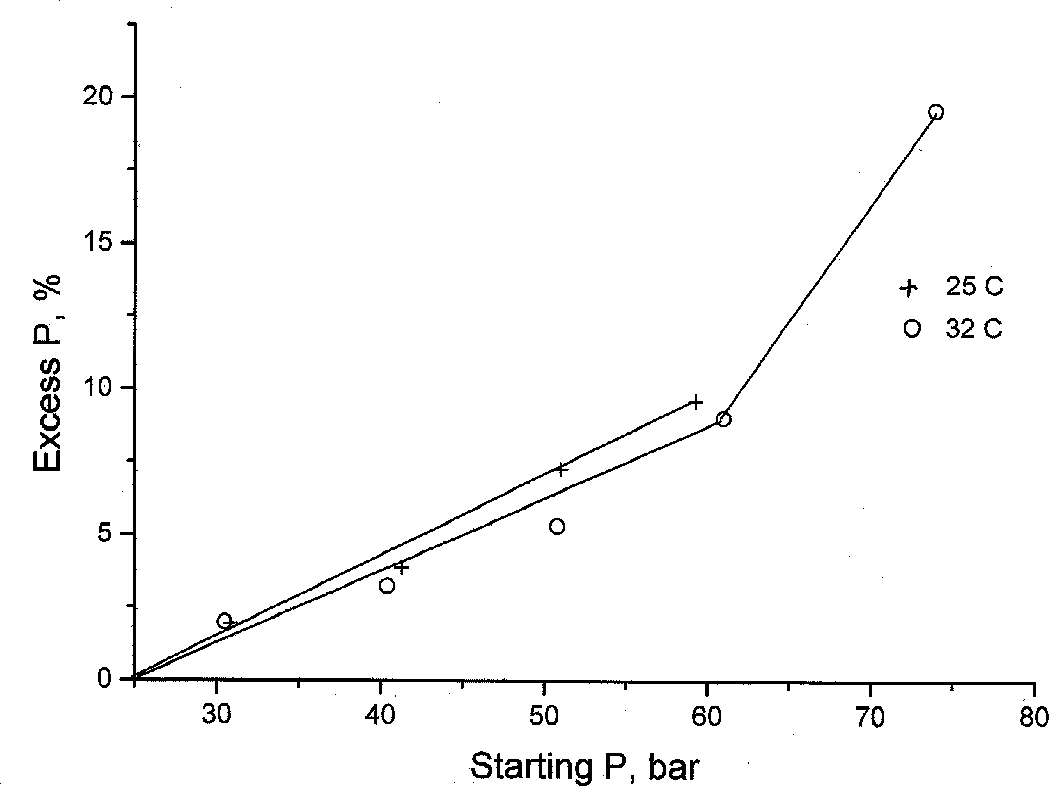
Fig.3. Dependence of the pressure gain in the process of the CO2 Ar interdiffusion at 25 and 32oC.
Gorbaty Yu.E. and Bondarenko G.V. The physical state of supercritical fluids.
It is consequential for practical goals to know, in what way the physical state of the supercritical solvent influences chemical processes in the system. So far, not much attention has been given to this topic, except rather timid attempts to classify the physical state of a supercritical fluid as liquid-like or gas-like, using density of the fluid as a criterion. Such a situation is understandable. From the theoretical viewpoint the only definition of the physical state is allowed: it is neither liquid, nor gas. Unfortunately, this negative statement is of no help for predicting the properties and behavior of supercritical fluids, which in addition to a great geological importance become now a competitive media for industrial technologies.
The goal of of our work was to analyze the temperature behavior of sulfur-saturated aqueous solutions and to suggest conditional criteria for classification of the physical state of high-temperature fluids. The system S - H2O makes a suitable model of a complex hydrothermal fluid containing electrolytes, non-electrolytes and the species, which at ambient conditions are gases. Therefore, inferences drawn from the behavior of this supercritical solution may be applied to many other supercritical systems.
The desire to understand the temperature trends for sulfur-bearing components in the high-temperature aqueous solution has led us to an assumption that the reason for the specific behavior of concentrations near the critical isotherm may be a qualitative change in the physical state of the fluid
40
at this temperature. However, to analyze the physical state of a supercritical phase we need to set conditional criteria defining the liquid and gaseous state. We suggested to use the probability of rotational molecular movement as the criterion. It was found that at supercritical pressures a noticeable part of the water molecule gains a possibility to rotate freely as temperature exceeds the critical value. Such a state of a supercritical fluid cannot be considered as liquid-like one but, on the other hand, it is far from being gas-like. The latter state is achieved at a density of about 0.1 g cm-3.. So, we had to introduce the concept of a transitory supercritical state that spreads over the whole range of temperatures and pressures of practical interest. It has been shown that the transitory supercritical phase is a unique state of a fluid characterized by strong structural fluctuations.
It is interesting that the increase in rotational freedom of solutes in diluted solutions is determined by the state of the matrix, that is, by a solvent. As can be seen in the work of Kieke et al. [19], the distinct rotational branches of CO2 molecules dissolved in water appear in the region of critical isotherm of water. It can hardly be expected that water molecules can rotate freely at so a high pressure as 517 bar and at so a low temperature as 110�C. However, they rotate wonderfully being dissolved in Xe (Bowman et al. [20]) which under these conditions is certainly in the supercritical state.
On the other hand, it is clear that the criteria suggested are not absolutely correct inasmuch as free rotation is possible in typical liquids, e.g., in liquid hydrogen, Ar, Xe, CH4, etc. Incidentally, from the theoretical viewpoint the whole problem discussed here is also incorrect. Still, it must be resolved for the benefit of interpretation of important geological processes and development of energy-efficient and environment-friendly technologies.
References:
Kalinichev A.G. Analysis of hydrogen bonding in water under hydrothermal conditions.
A detailed analysis of hydrogen bonding in water under hydrothermal conditions has been completed in 1997, based on the recently proposed hybrid distance-energy criterion of H-bonding. New Monte Carlo computer simulations were performed and good agreement is found with all available experimental data. With increasing temperature, the average number of H-bonds per a water molecule, <nHB>, decreases with the same slope for both high-density (~1.0 g/cm3) as well as low-density (~0.2 g/cm3) supercritical water, asymptotically approaching zero at higher temperatures and lower densities. Over the whole supercritical region, except for the most high-density states, <nHB> is always below the percolation threshold (~1.6) indicating that the continuous network of hydrogen bonds is broken. Nevertheless, even at the highest temperature and the lowest density simulated, some degree of hydrogen bonding is still present in the form of dimers and trimers. For supercritical conditions of 673 K and 0.66 g/cm3, average hydrogen bonds are almost 10% weaker, 5% longer, and about 8 degrees more bent, compared to those in normal liquid water. However, over 40% of them are still preserved in the supercritical state, in good agreement with estimates from all available experimental data.
Fig. 1. Temperature dependence of the average number of H-bonds per a water molecule, <nHB>, in water under hydrothermal conditions.

Reference:
41
Korzhinsky M.A., Tkachenko S.I., Zhdanov N.N., Bocharnikov R.E. Measurement of partial pressure of hydrogen and total gas pressure in a fumarole vent under monitoring conditions in the volcano Kudryaviy.
Information on change in the parameters of state and composition of fumarole gases in an intereruptive period suggests the idea of the dynamics of volcanic activity alteration and can be a criterion to predict volcanic eruptions. Most informative parameters in this respect are temperature of volcanic gas, its, pressure, and concentrations of such gas components as CO2, H2, SO2, and HCl. Now methods for a direct measurement in fumarole gas of said parameters (excluding temperature) are nearly absent. Information on the gases composition is generally obtained by sampling them to alkali-filled evacuated vessels with a subsequent analysis [1].
But such a method to study the dynamics of alteration of the volcanic gas state parameters is hardly acceptable due to the necessity of a regular gas sampling the conditions of which are, sometimes, difficult to reproduce (air admixing to the gas, different sampling and cooling rates, analytic errors in analysis). The field chromatography method employed in the recent times for a continuous measurement of the gas composition has also a number of disadvantages, the major one being the fact that during the investigations the composition of the cooled gas alters, i.e. quenching reactions occurring in it corrupt its composition.
In the year 1997 we have performed trial relatively long-period measurements of temperature, hydrogen partial pressure and the gas pressure in a volcanic channel in the volcano Kudryaviy in one of the high-temperature fumaroles. The results are reported in this work.
Methods and conditions of the measurements. The designs of the cell to measure a partial pressure of hydrogen (pH-cell) and a total pressure of the gas (PG-cell) in a fumarole vent are illustrated schematically in fig.1. The operation principle of the pH-cell is based on a selectively preferential hydrogen diffusion through a platinum membrane at elevated temperatures. The platinum membrane is hermetically connected via a metallic capillary with a sensitive pressure transducer the rear side of which is open to the atmospheric pressure. Inasmuch as at elevated temperatures hydrogen is capable of penetrating through platinum, at equilibrium its partial pressure in the gas equals its pressure inside the cell. The PG-cell is a quartz tube, connected through a silicon hose with a sensitive pressure transducer, the rear side of which is evacuated. In this version the cell measures gas pressure in the fumarole vent. This pressure is the sum of the excess pressure of the volcanic gas (Pg) and the atmospheric pressure (Pa), i.e. Pga = Pg + K.Pa, where K is the coefficient of the gas-dynamic resistance of the rock porous layer above the quartz tube end. In addition to these cells there is an atmospheric pressure cell (PH cell) that is a sensitive pressure transducer evacuated at one side. Standard tensoresistive transducers D-01 supplied by the Vniiteplopribor were used as sensitive pressure cells. Each cell is furnished with a power source, electron amplifier and temperature transducer which made it possible to increase the sensitivity range and to introduce the corresponding temperatural corrections. The cells were calibrated to a water U-shaped and the PH-cell was calibrated in the same manner to hydrogen values in equilibrium with the solid Fe3O4 - Fe2O3 , Ni-NiO, and Co-CoO buffers at one atmosphere of the water vapour at 800oC.
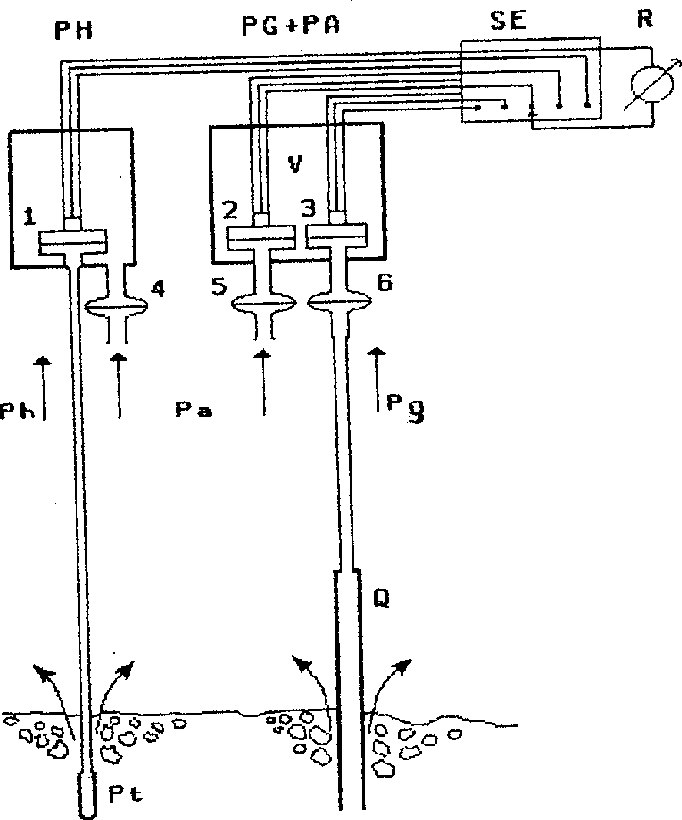
Fig.1. Principal scheme of cells for measurements of partial hydrogen pressure, gas pressure in fumarole vent and atmospheric pressures. PH-cell for measurements of partial hydrogen pressure; Pg + PA-double cell for measurements of gas pressure in fumarole vent and atmospheric pressures; SE-source of energy and switches; R-recorder of mV; V-vacuumed space; Pt-platinum membrane; Q-quartz tube; 1,2,3-pressure transducers; 4,5,6-protecting membranes.
Under field conditions the system was stationary installed at a fumarole of the volcano Kudryaviy (T=846oC) and the parameters in question were measured for 10 days in the daytime. The electric signals were registered with a millivoltmeter V7-41 by sampling sequentially the corresponding vents. The gas temperature in a fumarole vent was measured with a Pt-PtRd thermocouple with an accuracy of +10oC. The studies were performed from 4 to 13 of August 1997 in a relatively calm weather period. Initially oxygen fugacity was measured with an electrochemical sensor [2] and, episodically, the gas pressure-measuring cell was opened to the atmospheric pressure using a guide cock, which enabled us to measure the excess gas pressure directly on time.
Results. The diagram in fig.2. illustrates the change of the atmospheric pressure, total and excess gas pressures in the fumarole vent, and the partial hydrogen pressure with the time. The temperature of fumarole gases was constant to be 945+10oC in the process of the measurements. The atmospheric pressure varied from 0.939 to 0.916 atm, therewith at the background of its increase/decrease a regular 24-hour cycling was observed: it was maximum in early morning hours, and at 3-4 pm it passed the minimum. Episodic measurements of the excess fumarole volcanic gas pressure enable the calculation of the coefficient of the gas-dynamic resistance of the rock porous layer above the quartz tube end to be 0.997 . The excess gas pressure, calculated from the equ.:
42
Pg=Pga-K.Pa, varied from 0.0204 to 0.0085 in the process of the measurements and also showed a clear correlation with the atmospheric pressure.

Fig.2. Changes in atmospheric (Pa), total (Pga) and excess (Pg) pressures in fumarole vent and partial hydrogen pressure (Ph) with the time.
The change in the hydrogen pressure in the process of the measurements is of greatest interest. The partial pressure value varied from 0.0131 to 0.0062 in the process of the measurements and it also correlates with the changing atmospheric pressure. If the found dependence of the partial pressure of hydrogen on the atmospheric pressure is not an instrumental error, then the phenomenon can be explained by the supposition that the partitioning of various components (hydrogen in particular) between the heterogeneous phases (liquid-vapour, melt-gas) is highly pressure sensitive. Comparison of the measured hydrogen concentrations values with those derived from the gas analysis [2] shows their fair convergence within 0.8-1.1 mol% for this temperature. At the same time the hydrogen concentration values calculated from the measured oxygen fugacity value (logfO2=-12.8) have a somewhat lower value of 0.6 mol%. In work [3] it was shown that the measured oxygen fugacity values diminish with the decreasing temperature and obey the reaction SO2 +H2O = H2S +O2. As in the gas samples taken from high-temperature fumaroles the hydrogen concentration keeps high values, this fact suggests that the reaction H2 +O2 = H2O is quenched upon a rapid gas cooling, high H2 values being retained therewith, i.e. hydrogen concentrations in the gas are nonequilibrium.
The investigations performed have shown that the gas pressure in the fumarole vent, like hydrogen fugacity in it are atmospheric pressure dependent. Upon a rapid gas cooling hydrogen is quenched and keeps high concentrations corresponding to the gas sampling temperatures. The methods developed for a direct measurement of the volcanic gas state parameters, being properly modified, can be successfully used for long-period observations of the volcanic activity.
References:
Bocharnikov R.E. Experimental research of interaction of volcanic gas with some rock-forming and ore minerals.
The interaction of high-temperature volcanic gas with some rock-forming and ore minerals has been experimentally studied for the first time. The experiments were run under natural conditions (Kudriavy volcano, Kuril Islands). The interaction of gases with natural minerals, namely, albite, labrador, orthoclase, olivine, apatite, epidote, diopside, calcite, biotite, barite, and sphalerrite was studied at 620, 910oC under normal pressure. The runs with biotite were conducted only at 910oC, and with diopside and epidote only at 620oC. The duration of the runs was 650 h.
The microprobe analysis data suggest that:
-at 620oC apatite, diopside, epidote, olivine, and sphalerite are stable. Calcite is replaced by anhydrite, barite suffers either a thermally-assisted collapse or dissolves in the gas phase;
-at 910oC apatite is stable. Calcilte, barite, and sphalerite completely break-down or dissolve in the gas. The initial composition of biotite gets potassium-depleted and iron-enriched, the concentration of Fe being in direct correlation with the concentration of Mg and in inverse correlation with the concentration of Al;
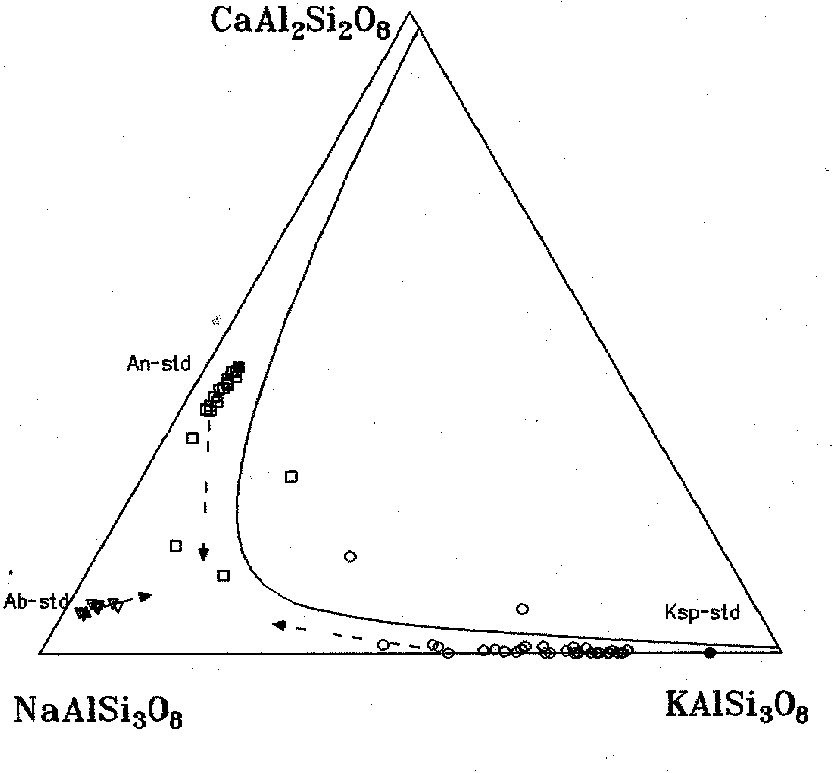
-the gas-olivine interaction at 910oC and the gas-feldspars interaction both at 910oC and 620oC is governed by cation - exchange reactions occurring by the diffusion mechanism. Mg in olivine is replaced by Fe, and alterations of feldspars compositions trend towards the formation of a ternary composition in a K-Na-Ca-feldsparic solid solution in equilibrium with the gas (fig.1). Potassium is the most mobile element in the system volcanic gas-feldspars;
-the width of alteration zones amounts to several hundreds of microns (for 650 h) in some minerals, suggesting a conclusion about a rather high (in geological time scale) rate of the interaction reactions.
So, the interaction between high-temperature volcanic gases and minerals can be an important factor affecting alterations of host rocks in near-surface magmatic systems.
Zonova I.A. Migration of fluid inclusions in quartz in thermogradient fields.
A series of runs have been concerned with the behavior of water-salt inclusions in quartz at different thermal gradients. Crystal of synthetic quartz (France) containing a considerable amount of fluid inclusions (1M NaOH, density 0.8 g/cm3) oriented along the 'c'-axis were placed into a thermogradient chamber at 275oC with temperature gradients 14 and 4 oC/cm and kept there for 55 days and 12 months, respectively. The solubility and the solubility temperatural coefficient of quartz in 1M NaOH at 250-270oC coincide with these values for the system quartz-water at 500-700oC and 5-10 kbar [1,2,3], and using an alkaline solution and low temperatures makes it possible to avoid decrepitation of inclusions.
43
The transport distance of fluid inclusions as a function of size in the direction of thermal gradient
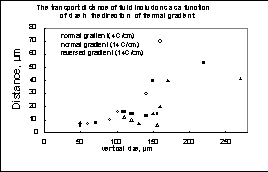
The migration rates of fluid inclusions measuring from 20 to 200 �m were estimated to be 1-5 m/day at 14oC/cm and 0.3-1 �m /day at 4oC/cm which at recalculation to the normal natural thermal gradient (30oC/cm) and the time period of one metamorphic event (about 10 mln years) yield a minimal migration route of inclusions 0.5-1.5 mm over 10 mln years (fig.1). The obtained result suggests that in thermogradient fields, which are universal in natural metamorphic systems, water-salt inclusions escape a crystal and migrate into the intercrystalline space, whereas a largest number of the rest of the carbonate inclusions represent not the general fluid composition but only a co-existing phase wherein the host-mineral dissolves insignificantly. This, again, supports the two-phase model of metamorthic fluid.
References:
Bocharnikov R.E., Korzhinsky M.A., and Tkachenko S.I. Intermixing of meteoric waters and a magmatic fluid in volcanic channels.
The process of intermixing of meteoric waters and a magmatic fluid in volcanic channels has been interpreted. The interpretation is based on the isotopic and chemical composition data obtained for volcanic gases of the volcano Kudriavii (Taran, 1995) and on the solution of a thermal problem of cold-hot water intermixing.
The solution of the thermal problem yields the dependence of the mixture temperature on the share of the cold and hot water under various pressures that fairly agrees with the dependence of the gas temperature on the share of meteoric and magmatic components obtained from the isotopic data on hydrogen (fig.2) . It has been found that meteoric water starts boiling at pressures of 4-10 bar, i.e. at a depth not greater than 100 meters from the surface.
Fig.1. Solvus isotherm and compositions of feldspars at 910oC and normal pressure (the arrows indicate the trends of initial compositions alterations)
44
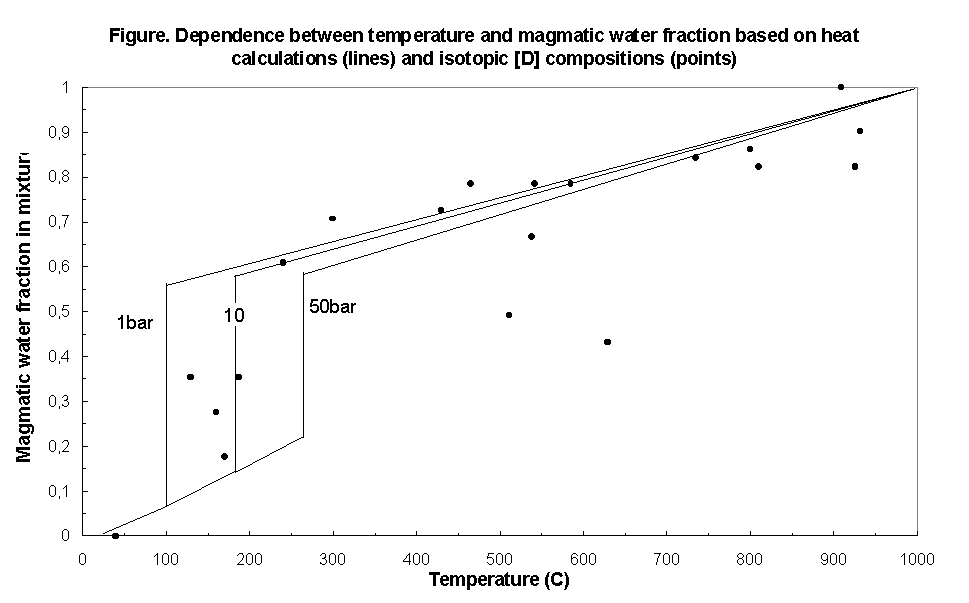
Fig.2. Dependence of the gas temperature on the share of the magmatic component in the mixture based on thermal calculations (lines) and hydrogen isotopic data (dots)
The temperature dependence of the share of the meteoric and magmatic components calculated from the chemical composition of the gases is inconsistent with the thermal intermixing balance. This inconsistency is possible due to the interaction of the high-temperature gases with the rock and/or due to an erratic gas composition at sampling.
Reference:
Budanov S.V. Solubility of diopside in an H2O-NaCl fluid at T=650oC and P=2-7.5 kbar.
Solubility of diopside single crystals in H2O-NaCl fluids (from 0 to 60 wt% NaCl) has been studied at 650oC and pressures 2.0 and 7.5 kbar. The 2 and 5 kbar runs were performed in hydrothermal set-ups of the exoclave type; the 7.5 kbar ones were performed on a gas bomb.
It has been found that at said parameters diopside dissolves incongruently with the formation of forsterite and some femic minerals (precise definitions are unavailable) on the crystal surface. A regular increase of the solubility of diopside with the growing pressure and NaCl concentration in the fluid has been found using a weight loss method and an analysis of quenched solutions. The dependence of the gross value of solubility (weight loss related to the fluid volume) on the concentration of NaCl in the fluid under different pressures is illustrated in fig.1. The dependence is linear. The gross value of solubility grows from 0 (2 kbar, pure water) to 4.88 g/l (7.5 kbar, 60 wt% NaCl). The dependence of Ca concentration in quenched solution on the fluid salinity is illustrated in fig.2. The concentration of Ca regularly increase from 10 ppm (2 kbar, pure water) to 1417 ppm (7.5 kbar, 60 wt% NaCl). As the fluid salinity grows the Si/Ca ratio grows insignificantly in a quenched solution.
A clear positive pressure and concentration dependence of solubility of diopside obtained in this work for the studied range of the parameters suggests a high stability of chloride complexes of Ca and SiO2 under the conditions of middle and lower crust. Low concentrations of Mg and Fe quenched solutions indicate that they stay on the compositions of neogenic minerals and the gross value of solubility is almost entirely due to SiO2 and Ca which transit into the solution. Allowing for the precipitation of quenched phases (wollastonite, to a smaller degree tremolite) one can conclude that real values of diopside solubility are somewhat higher than the cited ones. The results of the study are given at greater length elsewhere [1].
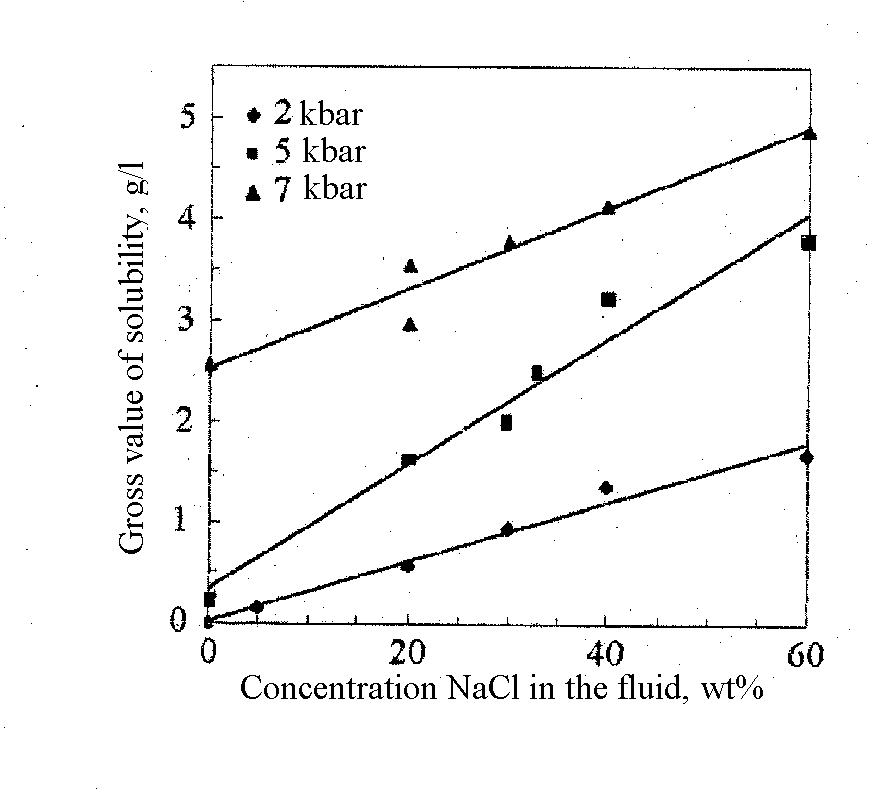
Fig.1. Gross value of diopside solubility Vs the fluid salinity at different pressures. Errors are within the size of the symbols.
45
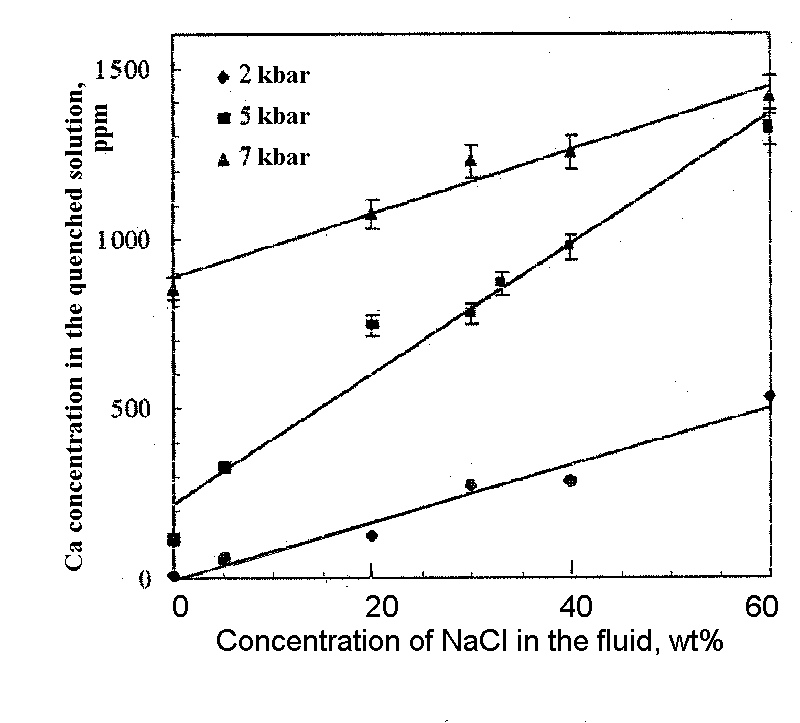
Fig.2. Ca concentration in the quenched solution vs the fluid salinity at different pressures.
Reference:
Budanov S.V., Schmulovich K.I. Experimental determination of diopside solubility in H2O-NaCl fluids at 650oC and pressures 2-7.5 kbar. // Geochim. To be publiched.
46