#Litvin Yu.A., Chudinovskikh L.T., Zharikov V.A., Shiryaev A.A., Galimov E.M., Saparin G.V., Obyden S.K., Chukichev M.V., Vavilov V.S. Diamonds of new carbonate-graphite syntheses: crystal growth in the Na2Mg(CO3)2 - K2Mg(CO3)2 - C system, carbon isotopy, scanning electron microscopy and cathodoluminescence.
key words [carbonate-diamond synthesis isotopy cathodoluminescence]
Crystallisation of diamond is firstly realised in the system Na2Mg2(CO3)2-K2Mg(CO3)2-graphite at 8 - 10 GPa and 1600 - 1800oC. Transparent colourless octahedral diamonds up to 0.15 mm in size were grown in conditions of spontaneous nucleation. Diamond seed growth was also carried out using cubo-octahedral single crystals of HP-synthetic "metal-carbon" diamonds (0.5 - 0.7 mm in size) as the seeds.
Carbon isotope analyses for starting materials and diamonds crystallised in the experimental carbonate - graphite systems are presented in the Table:
The isotopic data are in agreement with the concept of diamond crystal growth from carbon solutions in carbonate melts [2]. The calculated average 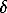 13C for starting mixtures is close to that for diamond, and this is a reason for a preliminary conclusion regarding a possibility of carbon isotopic exchange between carbonate and graphite carbon sources in the stage of melted state of diamond-forming system. The studies of the peculiarities of diamond crystal morphology by scanning electron microscopy shows that a preferable mechanism of diamond growth from the alkaline carbonate-carbon melts is a deposition of the newly grown diamond material by layers in parallel with the octahedral faces.
13C for starting mixtures is close to that for diamond, and this is a reason for a preliminary conclusion regarding a possibility of carbon isotopic exchange between carbonate and graphite carbon sources in the stage of melted state of diamond-forming system. The studies of the peculiarities of diamond crystal morphology by scanning electron microscopy shows that a preferable mechanism of diamond growth from the alkaline carbonate-carbon melts is a deposition of the newly grown diamond material by layers in parallel with the octahedral faces.
For diamonds grown in alkaline-carbonate melts, colour cathodoluminescence (CCL-SEM) patterns exhibit the lack of luminescence or the existence of red emission with extra low intensity for octahedral faces. The red emission intensity increases in the directions toward cubic pseudo faces of single crystals. Observed peculiarity of CCL-SEM emission is a specific property of synthetic "carbonate-graphite" diamonds in comparison with both natural and synthetic "metal-graphite" diamonds.
There are three major bands in the cathodoluminescence (CL) spectra of synthetic carbonate-carbon diamonds: (i) a broad nonstructural blue band within 445 - 480 nm (marked usually as the 'band A') having the maximal peak at 470 nm; (ii) a green band with the dominating system of the line 503 and its phonon replicas at 512.6 and 521 nm (H3 - system); (iii) a band system within 575 - 640 nm consisting of a zero-phonon line at 576.3 nm and its phonon replicas with phonon energy 40 - 3 MeV. CCL-SEM and CL-spectroscopy data are consistent with the version of belonging carbonate-carbon diamonds to the type IIa.
#Chudinovskikh L.T., Litvin Yu.A., Aldushin K.A. Experimental studies of diamond growth on the {111} and {100} faces of the seed crystals at 7-10 GPa.
key words [diamond high pressure experiment seed stimulated growth]
High-pressure spontaneous crystallization of diamond in the systems K2Mg(CO3)2-C (graphite) and Na2Mg(CO3)2-C was first reported in [1,2]. Transparent, practically colourless octahedral diamond single crystals up to 0.15 mm in size were formed. Physicochemical conditions of diamond crystallization were interpreted as a crystal growth from carbon solution in carbonate melt ('solution-melt method'). The system Na2Mg(CO3)2 - K2Mg(CO3)2-C was used for a seed stimulated diamond growth as well [3]. These processes are the results of further developing carbonate-carbon diamond synthesis originated by [4,5,1].
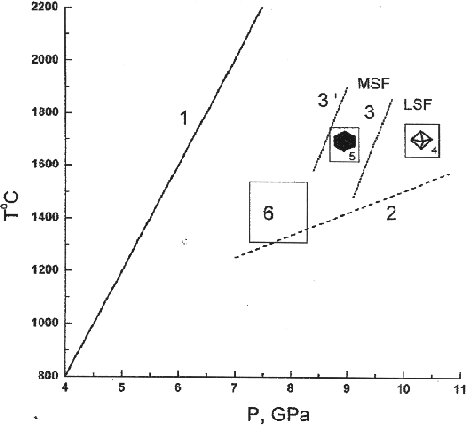
Some new data concerning crystallization of diamond in the Na2Mg(CO3)2-K2Mg(CO3)2-C system are presented in the paper. The experimental conditions are specified in fig.1 showing a lowering of the LSF-MSF boundary in the case of using the Na2Mg(CO3)2 solvent in comparison with the K2Mg(CO3)2 solvent. The spontaneous single crystals formed in the vicinity of the boundary in the Na2Mg(CO3)2 - C system are presented by the scanning electron microscopy (SEM) patterns in fig.2.
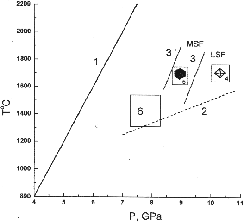
Fig.1.Conditions of high-pressure and high-temperature experiments. (1) graphite-diamond equilibrium curve, (2) approximate pressure dependence of the eutectic temperature of the K2Mg(CO3)2-graphite system, (3) boundary between the field of metastable supersaturation of carbon solution in the alkali-carbonate melt with respect to diamond (MSF) and the field of labile solutions (LSF), (4) runs with diamond crystallization, (5) runs with matastable crystallization of graphite single crystals, (6) runs with partial dissolution of diamond single crystals in the alkali-carbonate melt and the formation of the cubic K2Mg(CO3)2 variety, (3) boundary between the fields MSF and LSF for the system Na2Mg(CO3)2- graphite.
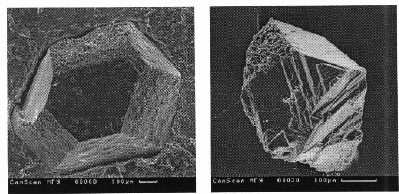
Diamond seed-stimulated growth was realised in the Na2Mg(CO3)2 - C and NaKMg(CO3)2-C systems close to the LSF-MSF boundary conditions. The seeds were cubic -octahedral diamond single crystals (about 0.5 mm in size) (Fig.3,a) synthesized in the �metallic� Mn-Ni-C system (Mn50Ni50, wt. % alloy was used). Natural diamonds from Yakutia were used as the seed crystals as well (Fig.3,b).
It is well known [6] that the synthetic 'metal-carbon' diamonds grow by layers in parallel with both octahedral and cubic faces forming the corresponding growth pyramids. This is a reason for optical and physical inhomogeneity of 'metal-carbon' diamonds. It was also found [6] that high quality natural diamonds grow just by the layers in parallel with octahedral faces.
# This work is supported by the Russian Foundation for Basic Research
(project N 96-05-64786 and N 98-05-64033) and Federal Programm "Integration" (project N 250).
50
Investigation of the peculiarities of diamond seed growth using scanning electron microscopy technique shows that newly formed layers grown over octahedral faces of the seed crystals are of the same octahedral orientation. As a result, smooth faces are developed with growth patterns presented by the irregular growth ridges and steps, vicinal forms, growth trigons, etc. (Fig.4). But, the newly grown layers over cubic faces are formed with intimately contacted octahedral microcrystals, and coarsely rugged face surfaces are developed (Fig.5). This clearly demonstrates that a preferable mechanism of diamond growth from the alkaline carbonate-carbon melts is a deposition of the newly grown diamond material by layers in parallel with the octahedral faces.
These peculiarities are typical for the growth mechanism of natural diamonds of kimberlite deposits.
References:
- Litvin Yu.A., Chudinovskikh L.T., Zharikov V.A. (1997)// Dokl. Ak. Nauk, V.355, N5, 669-672.
- Litvin Yu.A., Chudinovskikh L.T., Zharikov V.A. (1998)// Dokl. Ak. Nauk, V.359, N5, 668-670.
- Litvin Yu.A., Chudinovskikh L.T., Zharikov V.A. (1998)// Dokl. Ak. Nauk, V.359, N6, 818-820.
- Akaishi M. (1993)// Diamonds and Related Materials 2, 183-189.
- Taniguchi T.,Dobson D.,.Jones A.P., Rabe R., Milledge H.J. (1996) J.Mater.Res.,11,N10, 1-11.
- Shigley J.E., Fritsch E., Stockton C.M., Koivula J.I., Fryer C.W. Kane R.E., Hargett D.R., Welch C.W. (1987)// Gems and Gemology. 23, N4,187-206.

Fig.2. Spontaneous octahedral diamond crystals (Na2Mg(CO3)2 - C join, 9.5 GPa, 1800oC).

Fig.3.(a) synthetic 'metal-carbon' diamond seed overgrown with 'carbonate-carbon' diamond layers (NaKMg(CO3)2-C join, 10 GPa, 1700oC, 33 min.), (b) a piece of Yakutian natural diamond seed covered with 'carbonate-carbon' diamond after 25 min. treatment at 9 GPa, 1600oC (Na2Mg(CO3)2-C join).
51

Fig.4.Growth forms on octahedral face of synthetic seed crystal shown in fig.3(a).

Fig.5.(a) adjacent {111} and {100} faces of synthetic seed crystal shown in fig.3(a), (b) octahedral micropyramid growth forms on cubic face of the same crystal (Fig.3(a)).
Sokol A.G., Pal'yanov Yu.N., Borzdov Yu. M., and Shatskii A.F. Diamond crystallization in melts of alkaline carbonates at 7 GPa and 1700oC.
key words [diamond alkaline carbonates melt synthesis]
Institute of Mineralogy and Petrography, Siberian Branch, Russian Academy of Sciences, Novosibirsk
The problem of the medium of natural diamond formation is one of the main ones in the mantle petrology. The progress in the recent time on diamond crystallization in carbonate-carbon systems, their specific physicochemical properties, and a special role of carbonate melts in the mantle petrology put carbonate systems in a series of the most promising subjects for modeling of diamond genesis.
It is established in experiments with duration up to 30 min that spontaneous diamond nucleation in the "carbonate-graphite" systems occurs at either P = 7.7 GPa and T > 2000oC or P > 9.0 GPa and T > 1600oC [1-3]. The size of the crystals obtained is usually 10-20  m, seldom, 100 m.
m, seldom, 100 m.
Experiments on diamond crystallization were performed in the Na2CO3-C and K2CO3-C systems on a high-pressure pressless "split sphere" type BARS apparatus at P = 7 GPa and T = 1750-1700oC. The temperature was measured by a PtRh 30/6 thermocouple, whose junction was placed inside the heater. To determine the correction to e.m.f. of the thermocouple, a series of experiments was performed at 7.0 GPa in which the moment of melting of a nickel wire in the NaCl was detected by measuring the electric resistance. Since melts of alkaline carbonates, being low-viscous, are very mobile and reactive, a procedure providing experiments with duration up to 18 h was developed. The studies were performed in graphite and platinum ampules using schemes accepted for diamond growth in metal-carbon systems [4]. It is established that a graphite container does not provide sufficient leak-proofness of alkaline carbonate systems in prolonged experiments because of penetration of the carbonate melt via intergrain boundaries of graphite and decomposition of the wall due to synthesis of diamond in it [5]. The interaction of the carbonate melt with an isolating bush results in extraction of magnesium oxide and its combined crystallization with diamond. The entire leak-proofness of the crystallization volume of the system is provided only in the case when Pt ark-welded ampules are used.
In prolonged experiments using the Na2CO3-C and K2CO3-C systems, diamond crystallization was established in the following processes:
1. Diamond nucleation at the graphite-carbonate interface and further crystal growth through the carbonate melt film. The maximum size of diamond crystals is 600-700 m.
2. Diamond growth on seeding through the carbonate melt film separating graphite and seed crystal.
3. Spontaneous diamond nucleation in the carbonate melt bulk oversaturated with carbon.
4. Seed stimulated diamond growth from the carbon-saturated carbonate melt in the "cool" part of the crystallization ampoule. The maximum thickness of the diamond layer grown on seeding is 400-500 m.
Two different mechanisms of diamond crystallization in the carbonate melt are suggested: (1) through the carbonate film separating diamond from metastable graphite and (2) in the temperature gradient field from the carbon solution in the carbonate melt.
52
References:
- Akaishi M., Kanda H., Yamaoka S. (1990) Synthesis of diamond from graphite-carbonate systems under very high temperature and pressure. // J. Cryst. Growth. V.104, pp.578-581.
- Taniquchi T., Dobson D., Jones A.P., Rabe R., Milledge H.J. (1996) Synthesis of cubic diamond in the graphite-magnesium, carbonate and graphite-K2Mg(CO3)2 systems ar high pressure of 9-10 Gpa region. // J. Mater. Res., V.11, N.10., p.1-11.
- Litvin Yu. A. , Chudinovskikh L. T. , and Zharikov V. A. (1997) Crystallization of Diamond and Graphite in Mantle Alkaline-Carbonate Melts in Experiment at 7-11 Gpa. // Dokl. Akad. Nauk, V.335, N. 5, pp. 669-672 (in Russian).
- Pal'yanov Yu.N. , Khokhryakov A.F., Borzdov Yu.M., Sokol A.G., Gusev V. A. , Rylov G. M. , and Sobolev N.V. ( 1997) Conditions of Growth and Real Structure of Crystals of Synthetic Diamond. // Geologiya i Geofizika, V.38, N. 5, pp. 882-918 (in Russian).
- Sokol A.G., Pal'yanov Yu. N., Borzdov Yu. M., Khokhryakov A.F., and Sobolev N.V. (1998) Diamond Crystallization in Melt of Na2CO3. // Dokl. Akad. Nauk, V.360, N.3 (in press) (in Russian).
#Borzdov Yu. M., Pal'yanov Yu. N. Efremov A.V., Sokol A.G. and Khokhryakov A.F. Crystallization of diamond and graphite in the Ni-Fe-C-N system
key words [diamond graphite crystallization nitrogen]
Nitrogen is the main structural impurity in diamond, and its content in natural crystals reaches 0.5 at.%, which suggests a high concentration of nitrogen in natural diamond-forming systems. Experimental studies on diamond crystallization in systems with additives of nitrogen-containing compounds are restricted and related to synthesis of diamond from graphite only. It is shown in [1] that the introduction of the N admixture into the Ni0.4Mn0.6 system increases the activation energy of nucleation of diamond.
The experimental study of the effect of an increased concentration of nitrogen on specific features of carbon crystallization (diamond and graphite) by the temperature gradient method was performed in the basic Ni-Fe-C system on a BARS apparatus by the procedure described previously [2]. The increased concentration of nitrogen was achieved due to the addition of iron nitride Fe3N in amount of 1.6.10-5 to 1.6.10-1 wt.% to the growing system. The other parameters were maintained unchanged in the series of experiments: P = 5.5-0.6 GPa, T = 1400oC, t = 65 h, the scheme of the assembly is completely identical, seeding orientation by (111). The morphology of diamond crystals grown in control experiments without Fe3N additives is determined by the development of faces {111}, {100}, {311}, and {110} in a sequence of decreasing significance, the dislocation density is at the level of 10-102 cm-2, and single inclusions of tenite were established only in the bases of the crystals. When the weighed sample of nitride Fe3N (CFe3N) increases from 0 to 3.1.10-4, the number of microtwins and tenite inclusions increases in the diamond crystals. At CFe3N > 3.1.10-4 wt.%, the diamond crystals are characterized by both microtwinning and splitting to form subindividuals, whose disorientation angles reach 7o relative to (111). At CFe3N > 1.6.10-3 wt.%, aggregative crystals with a great number of macro- and microtwins and intense splitting of faces {111} are formed.
In the interval of CFe3N values from 1.6.10-3 to 1.3.10-2 wt.%, the intensity of twinning and splitting of aggregative crystals increases, faces {100} of subindividuals remain planar, and form {111} is characterized by the vicinal roughly laminated structure. No spontaneous nucleation was observed in all experiments with Fe3N additives. The morphology of diamond crystals is determined only by faces {111} and {100}, and the role of faces {111} increases as the concentration of Fe3N increases. The crystals with elements of cleavage and twinning and aggregative crystals are characterized by the specific internal structure and high interference colors (to red) in the polarized light.
At CFe3N = 1.3.10-2 wt.%, in the cool zone of the crystallization ampule, the appearance of recrystallized graphite was observed along with the diamond aggregate fixed on seed. In the 1.6.10-1 > CFe3N > 1.3.10-2 wt.% range, diamond is formed by neither the synthesis method nor temperature gradient method. The starting graphite is dissolved in metal and crystallized in the cool zone as an aggregate of cleaved graphite crystals. A similar effect has been previously observed for diamond crystallization in metal-carbon systems with additives of H2O [3].
Conclusions
1. In the Ni-Fe-C-N system studied, as the concentration of the Fe3N admixture increases, the diamond crystals loose the morphological stability in the following sequence: single crystal --> crystal with microtwins --> aggregative crystal --> aggregate, and the intensity of twinning and cleavage increases.
2. When the concentration of Fe3N is higher than 1.3.10-2 wt.%, in the Ni-Fe-C-N system the nucleation and growth of diamond are completely ceased, and graphite is crystallized in the thermodynamic stability field.
The authors are grateful to M. Yu. Mikhailov for synthesis of nitride Fe3N.
References:
- M. I. Samoilovich, N. G. Sanzharlinskii, and V. I. Zadneprovskii, (1987) Physicochemical Parameters and Their Role in Diamond Crystallization in the Metal Melt-Carbon System, in Synthesis of Minerals, Moscow: Nedra, , pp. 336-367 (in Russian).
- Paly'anov Yu.N., Khokhriakov A.F., Borzdov Yu.M., Sokol A.G. (1995) On the role of water in growth and dissolution of diamond// 8-th International Simposium on water-rock interaction. Vladivostolk., Eds. Y.K.Kharaka, O.V.Chudaev . Rotterdam, Balkema, pp.95-98.
- Yu. N. Pal'yanov, A. F. Khokhryakov, Yu. M. Borzdov, A. G. Sokol, V. A. Gusev, G. M. Rylov, and N. V. Sobolev, (1997) Conditions of Growth and Real Structure of Synthetic Diamond Crystals, Geologiya i Geofizika,, vol. 38, no. 5, pp. 882-918 (in Russian).
# This work was supported by the Russian Foundation for Basic Research (project N 97-05-65195)
53
#Yakovlev E.N., Voronov O.A., and Rakhmanina A.V. Diamond synthesis in the carbon-hydrocarbon system at high pressure .
key words [diamond synthesis hydrocarbon]
L. F. Vereshchagin Institute of High-Pressure Physics, Russian Academy of Sciences, Troitsk, 142092 Moscow Region
The hypothesis of organic origin of diamonds (at least, a part of diamonds) [1, 2] stimulates studying systems containing mixtures of carbon materials and hydrocarbons. Experiments at high pressures with mixtures of these substances were carried out to elucidate the role of hydrogen in hydrocarbons in the transformation of carbon into diamond.
It is known that the "direct transition," i.e., transformation of graphite into diamond without catalyst occurs at pressures not lower than 10 GPa and temperatures ~2000oC. The transformations of other carbon materials into diamond are more difficult if any.
Experiments with mixtures were carried out by a standard scheme [3, 4]. High-pressure cells of the "toroid" type with the hole diameter of 15 mm were used. A reaction mixture in the form of a pellet ~60 mm3 in volume was placed in a cylindrical graphite heater. The mixture was prepared of the powder of one of the carbon materials: graphite MG-OSCh, graphite PG-6, carbon fibers or cokes of different trade marks and one of hydrocarbons: naphthalene, anthracene, adamantane, diphenyl, paraffin, black coal pitches, and others.
The carbon materials and hydrocarbons represent substances with different bonds: sp, sp2, sp3, and mixed bonds.
After the mixtures were treated under pressures of 8.0 GPa and temperatures to 1700oC for 10-30 s, the following facts were observed:
1. The formation of diamond occurs if the content of hydrogen in the mixture is not lower than 0.3 wt.%. If the content of hydrogen in the mixture is higher than 1 wt.%, all the carbon of the mixture in the reaction zone can be transformed into diamond.
2. The formation of graphite occurs at 700-1200oC.
3. The formation of diamond occurs at 1200-1500oC;
at T = 1500oC, all the carbon of the mixture in the reaction zone is transformed into diamond.
The experimental results are almost independent of the specific type of carbon and hydrocarbon from the list presented above.
It is noteworthy that there is a region of P, T parameters where graphite is formed in the region of the thermodynamic stability of diamond.
The results obtained suggest the following:
I. Hydrogen (or, strictly speaking, hydrogen-containing products of thermal destruction of hydrocarbons) in hydrocarbon facilitates the transformation of carbon into its modification of diamond, which is stable at high pressures.
II. The "special" action of hydrogen occurs at T >= 1200oC, i.e., under conditions of the appearance of atomic hydrogen.
III. The experimental results are independent of the type of bonds between atoms in carbon (sp2 in graphite, sp3 in carbon fibers, and sp, sp2, and sp3 in coke) and the type of bonds between carbon and hydrogen atoms in hydrocarbons (sp2 in aromatic hydrocarbons, sp3 in adamantane), which can be due to the formation of diamond through an intermediate reaction or activated complex, whose appearance in the first approximation is slightly sensitive to the type of the bond.
References:
- Vasil'ev V. G. , Koval'skii V. P. , and Cherskii N. V. Origin of Diamond, Moscow: Nedra, 1968, 216 pp. (in Russian).
- Orlov Yu. L. Mineralogy of Diamond, Moscow: Nauka, 1973, 221 pp. (in Russian).
- Yakovlev E.N., Voronov O.A., and Rakhmanina A.V. Synthesis of Diamonds from Hydrocarbons, Superhard Materials, 1984, no. 4, pp. 8-11 (in Russian).
- Yakovlev E. N., Voronov O. A., and Rakhmanina A. V. Polycrystalline Diamonds Materials Prepared Using Hydrocarbons, Superhard Materials, 1987, no. 2, pp. 3-51 (in Russian).
Feldman V.I., Sazonova L.V., Kozlov E.A., Zhugin Yu.N., Litvinov B.V. Experimental study of transformation of minerals under spherically convergent shock waves.
key words [mineral transformation shock wave]
Lomonosov Moscow State University, 119899 Department of Petrology; Russian Federal Nuclear Center All-Russian Research Institute of Engineering Physics 456770 Snekhinsk Tschelyabinsk distr., Russia
A new method of shock wave loading of rocks (developed in the All-Russian Research Institute of Engineering Physics) [1-4] enables one to study the behaviour of substance under loadings to 1000 GPa and at temperatures to 75.103 K. An investigation of shock-assisted metamorphism of pegmatite, granite, enstatite, wollastonitic and plagioclase garnet rocks was performed in spheric samples (dia about 5.0 cm). The pressures at the external sphere surface were 20-30 GPa whereas at the radius R=1 mm they amount to 250 GPa. Samples of pegmatite and granite demonstrate in the solid-phase transformation region (at stresses to 50 GPa) [5,6] a countermigration of cations at the feldspars contact: K-to plagioclase, Na and, to a smaller degree, Ca- to microcline. As a result of this process there arises a region of near-contact transformation of which the width (L, m) is directly proportional to the stress amplitude ( xx, GPa) and amounts to 300 m. The connection between these two values is different for pegmatite (xx = 20.7 + 0.0977L) and granite (xx = 20.7 + 0.137L) which is due to different calcium content of plagioclase (An3 and An7) respectively.The analogous equations (xx = 20.7 + 0.0977L) were obtained for impacted garnet-biotite gneisses in the astroblem Popigay (at the initial plagioclase An29). These three equations were used as the basis of the program for the calculation of shock loading on a rock sample.
xx, GPa) and amounts to 300 m. The connection between these two values is different for pegmatite (xx = 20.7 + 0.0977L) and granite (xx = 20.7 + 0.137L) which is due to different calcium content of plagioclase (An3 and An7) respectively.The analogous equations (xx = 20.7 + 0.0977L) were obtained for impacted garnet-biotite gneisses in the astroblem Popigay (at the initial plagioclase An29). These three equations were used as the basis of the program for the calculation of shock loading on a rock sample.
# This work was supported by the Russian Foundation for Basic Research (project N 97-05-64576)
54
The compression of enstatite, wollastonite [4], and garnet-plagioclase-hedenbergite spheres [7,8] in spherically convergent shock waves makes it possible to reveal in them the zones of characteristic physical-chemical transformations. In the external zones of the compressed spheres the minerals exhibit numerous planar cracks whose number grows in a unit volume as the center is approached. In deeper zones lying along the radius there begin high-temperature and high-pressure transformations of the substance. The low-temperature  -wollastonite recrystallizes that is accompanied by its inversion to the high-temperature
-wollastonite recrystallizes that is accompanied by its inversion to the high-temperature  -wollastonite [4]. Individual enstatite and hedenbergite grains [7,8] exhibit the onset of iron migration. Hedenbergite exhibits a decrease of the 2V angle to 50o as compared to 62o of unaltered pyroxenes. Some hedenbergite grains acquire a plate-like structure resembling the decay structure. The plates evolve in one or two directions, in the latter case they intersect at angles of 67 and 45o. The orientation of the plates intersecting at 67o is close to the orientation (001) and (100) faces. The compositions of different plates differ in concentrations of iron and magnesium. So, in this case redistribution of iron and magnesium takes place here that used to proceed in strictly definite crystallographic directions. In the same zone pyroxenes start melting with a subsequent escape of first iron and then magnesium. In the next zone, lying close to the center, at the front of spherically convergent shock wave the substance starts melting and boiling as the shock-compressed melt is being unloaded. Minute high-temperature phases crystallize from the melt. High-temperature cyclic -wollastonite crystallizes from the wollastonite melt; magnesium-rich pyroxene crystallizes from the hedenbergite melt. Some crystals are likely to form from superheated vapour.
-wollastonite [4]. Individual enstatite and hedenbergite grains [7,8] exhibit the onset of iron migration. Hedenbergite exhibits a decrease of the 2V angle to 50o as compared to 62o of unaltered pyroxenes. Some hedenbergite grains acquire a plate-like structure resembling the decay structure. The plates evolve in one or two directions, in the latter case they intersect at angles of 67 and 45o. The orientation of the plates intersecting at 67o is close to the orientation (001) and (100) faces. The compositions of different plates differ in concentrations of iron and magnesium. So, in this case redistribution of iron and magnesium takes place here that used to proceed in strictly definite crystallographic directions. In the same zone pyroxenes start melting with a subsequent escape of first iron and then magnesium. In the next zone, lying close to the center, at the front of spherically convergent shock wave the substance starts melting and boiling as the shock-compressed melt is being unloaded. Minute high-temperature phases crystallize from the melt. High-temperature cyclic -wollastonite crystallizes from the wollastonite melt; magnesium-rich pyroxene crystallizes from the hedenbergite melt. Some crystals are likely to form from superheated vapour.
References:
- Kozlov E.A. (1996) // In: Metals and minerals research in spherical shock-wave recovery experiments. Litvinov B.V. (ed.) Russian federal Nuclear Center-Research Institute of Technical Physics, Snezhinsk.
- Litvinov B.V. et al. (1991) // DAN V.319, N.6, pp.1428-1429.
- Kozlov E.A. et al. (1995) // Chim. Phys., V.14, N.1, pp.108-118.
- Kozlov E.A. et al. (1997) // DAN V.335, pp.328-332.
- Feldman V. et al. (1998) // Abstr. XXIX LPSC.
- Feldman V. et al. (1997) // Abstr. XXVIII LPSC.
- Sazonova et al. (1995) // Experiment in GeoSciences.
- Sazonova et al. (1997) //DAN, V.357, pp.395-398.
Goryanov S.V. Sidel'nikova Yu.M., Fursenko B.A., Belitsky I.A. Raman spectroscopy of phase transitions in dehydrated analcime under high pressures.
key words [analcime phase transition]
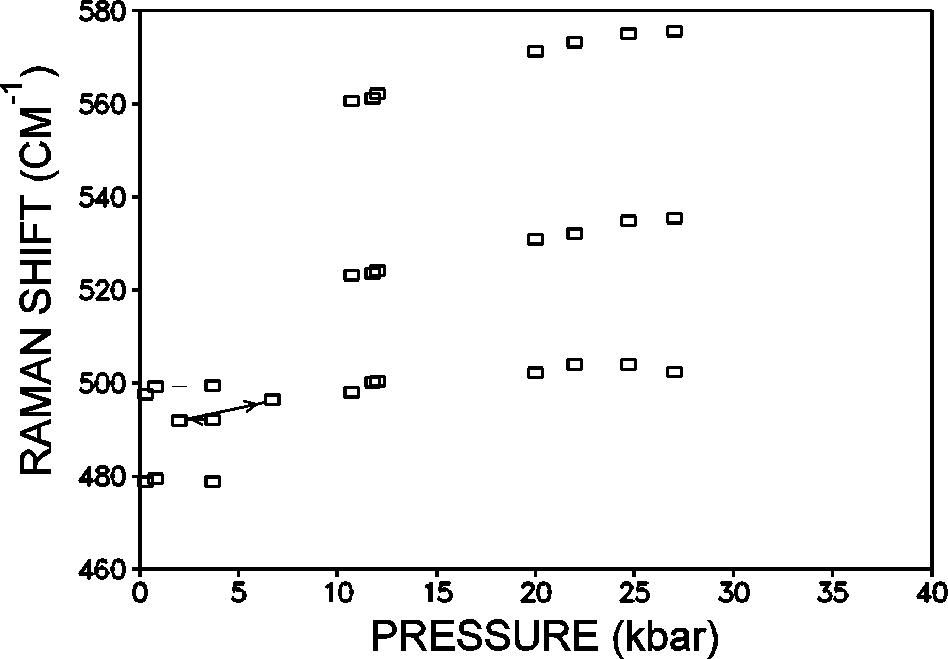
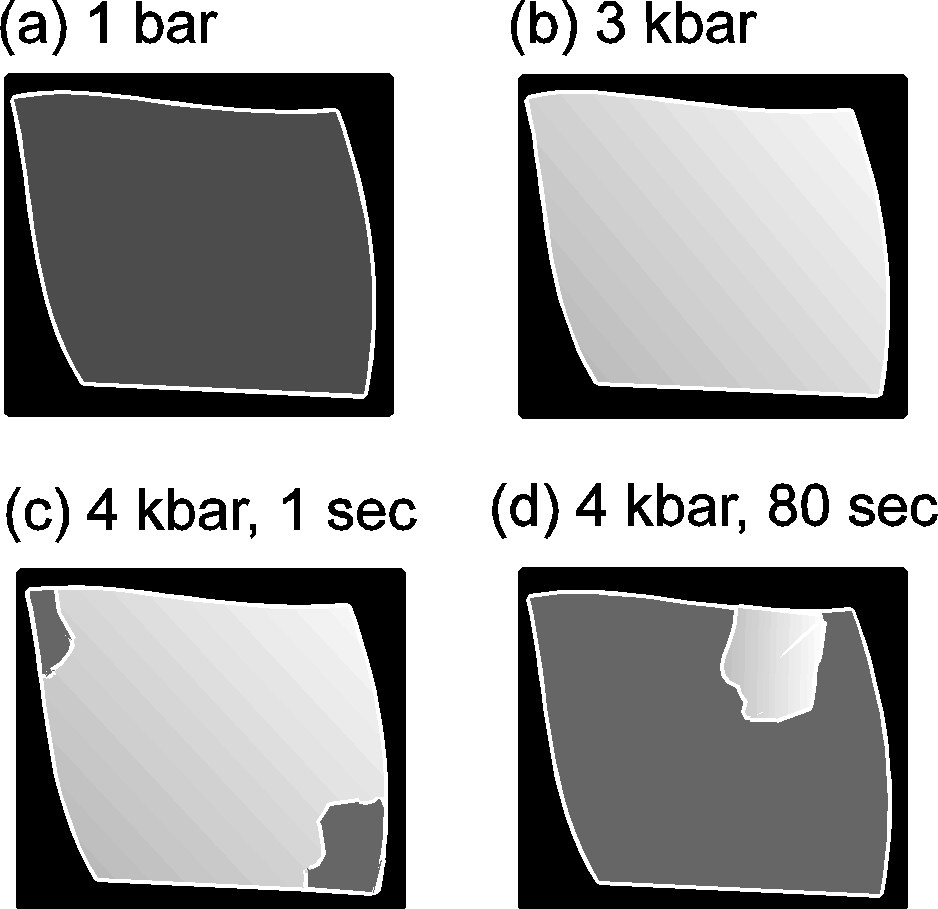
Phase transitions (PhT) and amorphization of alumosilicates under a pressure have been extensively studied recently [1-4]. Zeolites of the analcime group Na2[Al2Si4O12]2H2O have the framework of identical type and differ in the composition and order of tetrahedral cations T=Al, Si as well as in the filling of cavities with cations and water. Earlier Hasen and Finger [5] observed by X-raying four phase transitions (below 30 kbar) in natural analcime (Golden, Colorado). Water molecules in analcime channels hinder (by increasing the PhT pressure), complicate and blur the PhT, it is, therefore, of interest to primarily investigate transitions in dehydrated analcime which are accompanied by contrast zoning in accord with the preliminary data [2]. The investigation was performed using diamond anvils under a high hydrostatic pressure (P) of the medium (water-ices VI,VII or glycerin) to 37 kbar. Raman microspectroscopy (DILOR, OMARS89) [3] and polarizing microscopy (C. Zeiss) were used. Dehydrated monoclinic analcime (Nydym) demonstrates two phase transitions at 4 and 11 kbar (fig.1) each of which is reversible with a considerable hysteresis of =2 kbar. Several runs were performed for different samples, both water-compressed and glycerin-compressed, with increasing and decreasing P. Normally, analcime samples are twinned, and, in such crystals additional twinning arises under phase transitions and under an ice VI- to VII transition. A most clear pattern of phase transitions was obtained for an untwined crystal that exhibited a complete extinction at 1 bar (fig.2a). An observation in crossed polarizers showed a gradual lightening of the crystal (phase I crystal) with a pressure growth to 4 kbar (fig.2b). Then in the transition point at 4 kbar there appears a contrast dark zone of new phase II growing from the edges towards the crystal center (fig. 2c,d). In this case one can observe a nonuniform migration of the interphase boundary with a gradual deceleration. Phase II is observable visually and by the jump in the P-dependencies of the Raman spectra frequencies. The initial 480, 500 cm-1 doublet of strong Raman bands of the O-T-O oscillations (T=Al, Si) transforms during the PhT I-II to a shifted singlet that at PhT II-III is replaced by a triplet of bands.
The PhTs of the dehydrated analcime were observed at the same pressures of glycerin and water media. During the PhT water molecules do not penetrate into zeolite channels. As the sample was compressed in water-ices at 37 kbar, the Raman spectra demonstrated the entrance of a small amount of water (=8% of the water occurring in natural analcime) into the channels.
The disturbance of the crystal extinction with pressure is due to rotations of optical axes by different angles as stressed inhomogeneous deformations of the crystal arise.
The new high pressure phase eliminates these macroscopic stresses. Transitions in the analcime structure representable as superimosed 4-member rings of 4 tetrahedra are supposedly related to microdeformation of 4-member rings. Their deformation should be accompanied by deformation of TO4 tetrahedra and by the crystal symmetry change from monoclinic to triclinic.
55
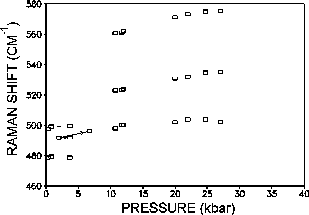
Fig.1. Dependence of the Raman frequencies of the O-T-O modes of dehydrated analcime, compressed in glycerin on the pressure.
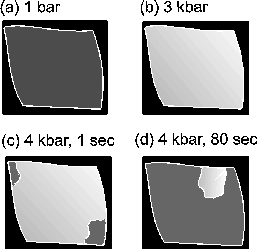

Fig.2. Dehydrated analcime, compressed in glycerin, observable in crossed polarizers under a microscope. The single crystal section is 180.150 mkm2. Schematic presentation of the major effect: (a) complete extinction of phase I at 1 bar; (b) gradual lightening of phase I with the growing pressure to 3 kbar; (c), (d) phase transition at 4 kbar: the migration of the interphase boundary for the time period of 1s and 80 s (dark zones with good extinction phase II, light part phase I); (e) photograph of the sample at 4 kbar, 60 sec later (dark edges brass plate, spotted zones with good extinction phase II, white zone phase I).
References:
- Tse J.S., Klug D.D., Ripmeester J.A., Desgreniers S., Lagarec K. // Nature. 1994. V. 369. P. 724-727.
- Goryainov S.V., Fursenko B.A., Belitsky I.A. // Phys. Chem. Minerals. 1996. V. 23. P. 297-298.
- Goryainov S.V., Belitsky I.A. // Phys. Chem. Minerals. 1995. V. 22, P. 443-452.
- Goryainov S.V., Fursenko B.A., Belitsky I.A., Kholdeev O.V.// Proceeding XIIIth Inter. Conf. Raman. Spectroscopy (Eds. W. Kiefer et al.). Wiley, Chichester, P. 918-919.
- Hazen R.M., Finger L.W. // Phase Transitions. 1979. V. 1. P. 1-22.
Leonova M.E., Kulinich S.A., Sevast'yanova L.G., Gulish O.K., Kravchenko O.V., Burdina K.P., Semenenko K.N. Synthesis of sodium bismuthide Na3Bi under a high pressure.
key words [sodium bismuthide high pressure] MGU, Chemical Departmant
We have investigated binary compounds of alkali metals involving elements of the principal subgroup of group V because at the formation of these compounds one can most clearly see a decrease in the molar volume of a synthesis product as compared to the gross volume of simple substances-constituents.
Pnictides of alkali metals of the type A3B possess the highest symmetry all the known pnictides of the general formula A3B crystallize either in hexagonal or cubic structure.
We have attempted to compare a volume reduction in the reactants at the compound formation and the compressibility of the substances under an external pressure for antimonides and bismuthides of alkali metals. The pressure required for a volume reduction of simple reactants to the A3B molar volume was calculated using the isothermal loading formula where  p is the pressure required for a volume change to occur, V/V is the change in the volume of the reactants, Vm is the molar volume of A3B, χ is the compressibility of the initial substances mixture.
p is the pressure required for a volume change to occur, V/V is the change in the volume of the reactants, Vm is the molar volume of A3B, χ is the compressibility of the initial substances mixture.
p=(V/V)/X
The compressibility of the substances mixture was calculated under the assumption of additivity of compressibility of individual substances at room temperature.
In this work we studied synthesis of sodium bismuthide from metallic sodium and bismuth in the temperature range 25-900oC both under normal and elevated pressures to 80 kbar, and in liquid ammonia at temperatures to 80oC.
The samples were treated in a high pressure cell using a standard 'toroid' technique. (The sample was placed into a self-sealing Ta capsule preventing the exchange of the substances inside and outside of the reacting volume).
The synthesis products were examined by the X-ray diffraction method (XRD).
In the temperature range 200-900oC the reaction proceeds to completion for 15 min both in the absence of the pressure and under pressures to 80 kbar. Sodium bismuthide also easily forms in liquid ammonia but in this case bismuth ought to be preamalgamated. It should be noted that thus produced Na3Bi exhibits an extremely high reactability.
56
An important result is the fact that in order to obtain Na3Bi it is sufficient to employ only pressure either static beginning with 3.5 kbar or short-time mechanic (grinding and intermixing of Na and Bi mixture), but in the absence of heating the reaction does not go to completion.
Orlov A.I., Khvostantsev L.G. Metastable phases of bismuth telluride of high hydrostatic pressure.
key words [bismuth telluride phase high pressure]
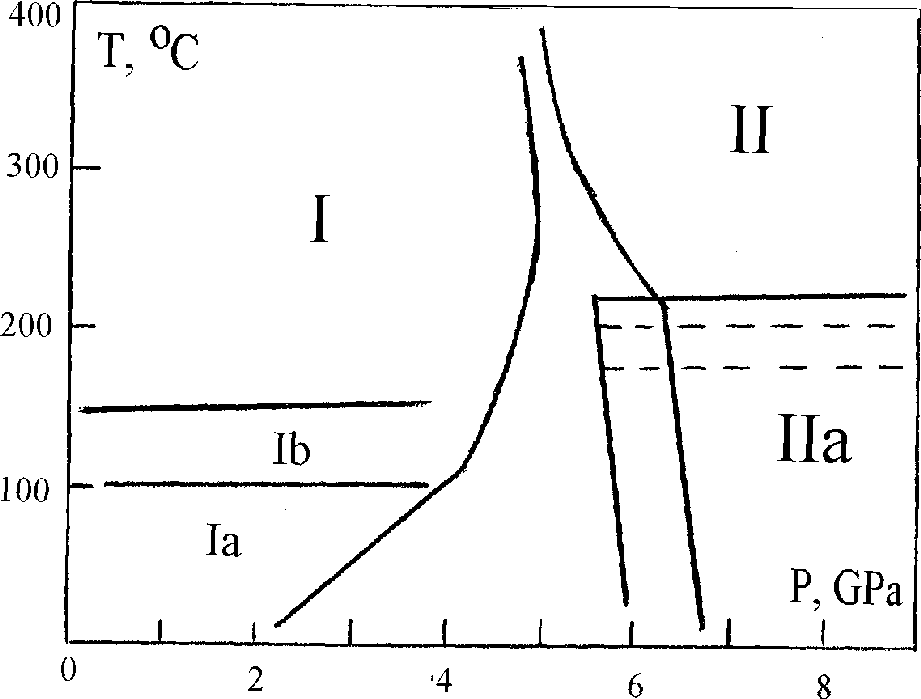
At pressures above 6 GPa under hydrostatic conditions in bismuth telluride the stable phase is phase II. It forms from the initial phase I at temperatures above 200oC trough an intermediate metastable phase. During pressure release high pressure phase II transforms to the initial one through two intermediate phases that can be metastable under normal conditions [I].
This work is dedicated to the investigation of formation peculiarities of metastable phases of bismuth tellurid. Techniques of precise measurements of electroresistance and thermo e.m.f. at hydrostatic conditions were used. The pressure was generated in the chamber of 'TOROID' type.
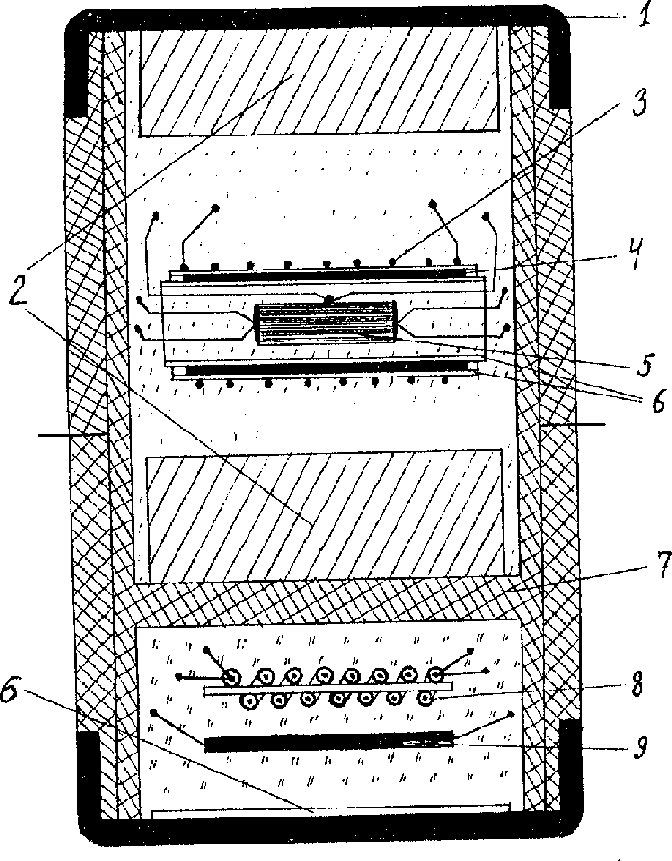
The precision of pressure and temperature measurements was provided by using a special high pressure cell (Fig.1):
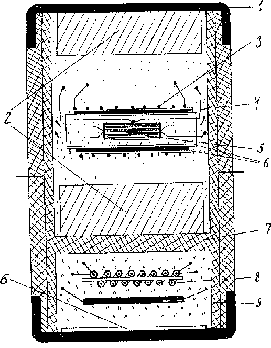
It consists of a compound capsule, made of fluoroplastics (7), hermetically sealed on top and bottom by copper covers (1). The sell consists of 2 hermetically separated parts that are filled with independent liquids to provide hydrostatic conditions.
The sample with electroimputs and thermocouple is located in the upper part of the cell inside cylinder heater (3). The copper screen (4) equalizes the temperature along the sample. Asbestos gaskets are used as heat insulator. The pressure sensor consisting of manganite resistance coil (8) and bismuth fine wire (9) is located in the lower part of the cell. The mica gaskets are electroinsulators.
The hermetic wall (7) between upper and lower parts is the distinctive detail of this cell which allows to fill them up with different liquids depending on the aim of the experiment. For example in this work the lower part was filled with methanol-ethanol mixture that provides hydrostatic pressure up to 10 GPa [3]. But this mixture decomposes under high temperature. That's why the upper part is filled with more heatstable petroleum ether [4] Phase Ia (Fig.2) has crystal structure R 3m [5] (PhaseI- R3m):
Under normal conditions it preserves up to 3 months. And as we have found out , it transforms to the initial phase through one more intrmediate phase - Ib. The phase Ib is a mixture of a crystal metastable phase and amorphous one.
The presence of the amorphous phase is registered by the special diffraction patterns of bismuth telluride samples. Besides the stoichiometric bismuth telluride samples of p-type the samples obtained by self-alloying (p-type) and dopirating by admixture SbJ (n-type) were studied. They showed that in these cases the temperature of the transition from metastable phase IIa to phase II decreases essentially (down to 150oC).
Thus, bismuth telluride has at least 3 metastable phases besides stable phases I and II. These metastable phases have no stability field on the P - T diagram and they are intermediate at I - II phase transitions.
References:
- Orlov A.I., Khvostantsev L.G. (1993) // Exp. In GeoSci., V.2, N.2, p.27.
- Khvostantsev L.G., Vereshchagin L.F., Novikov A.P. (1977) // High Temp.-High Press., V.9,N.6,p.637.
- Piermarini G.J., Block S., Barnett J.D. (1973) // J. Appl. Phys., V.44,p.5277.
- Barnett J.D., Bosco C.D. (1969) // J. Appl. Phys., V.40,p.3144.
- Atabaeva E.Ya., Itskevich E.S., Mashkov S.A., Popova S.V., Vereshchagin L.F. (1968) // S.S.F., V.10, N.1, p.62 (in Russian)
57
Kupin Yu.G., Rusakov V.S., Badyukov D.D., Pershin S.V. Impact induced change in the state of iron atoms: experiments with silicate-kamacite mixtures.
key words [silicate-kamacite mixture impact reaction]
Physics Department, Moscow, GEOKHI, N.N. Semenov Institute of Chemical Physics RAS, Chernogolovka, Russia.
Impact-induced reactions can be an important factor affecting the evolution of the protoplanet matter at the planet formation stage when the rates of the accreted matter fall onto the growing planet begin to reach the values of the order of several kilometres per second. The interaction of metallic iron with silicates can be one of such impact-induced reactions having a petrologic importance. In this work the occurrence of the metal-silicate reaction was confirmed by the Mossbauer spectroscopy examination of the products of the shock-wave experiments.
Powder mixtures of kamacite (5.87 wt% Ni, 0.4 wt% Co) with quartz, albite, oligoclase, enstatite, and olivine were subjected to the shock-wave action. The experiments were run in cylindrical containers that enabled us to realise the peak shock pressure from 80 to 90 Gpa in the axial target part. The studied matter was the material of the predominantly impact-induced melting zone consisting of glass (for feldspars, quartz and, partly, enstatite) or neogenic olivine with numerous inclusions of metallic spheerules. In addition, initial specimens of kamacite, enstatite, and olivine were studied.
The Mossbauer studies were carried out at room temperature on an electrodynamic spectrometer operating in constant acceleration mode. As the investigated specimens were locally inhomogeneous systems due to the occurrence of a large number of non-equivalent positions of iron atoms; the spectra were treated and analysed using the restoration of several independent distribution functions of hyperfine parameters, namely, hyperfine magnetic fields p(Hm), quadrupole shifts p(), and shifts p() of the components of the partial spectra.
Fig. 1 illustrates the spectra of the initial kamacite and the kamacite-quartz mixture under the action of shock waves. The spectrum of kamacite is a superposition of a great number of Zeeman six lines with somewhat different values of the field Hn due to the presence of nickel and cobalt atoms in the first coordination sphere of iron atoms. The restored distribution function p(Hn) of the hyperfine magnetic field is given in fig.2. As the result of the shock wave action on the kamacite-quartz mixture its spectrum exhibits the occurrence of two new partial spectra (fig. 1), namely a quadrupole doublet with the hyperfine parameters (` = 1.04 +-0.01 mm/s and `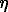 = 0.99+ 0/01 mm/s), characteristic of Fe2+ ions in a high-spin state, and a singlet with the shift ' = 0.05+0.03 mm/s characteristic of a Fe atom in the metallic state. Besides, the distribution function p(Hn), corresponding to the partial spectrum of kamacite in the mixture, in addition to the principal maximum at Hn337 kOe, has a local maximum at Hn305 kOe (fig.2). The presence of this maximum evidences for the appearance of a silicon atom in the closest neighbourhood of the Fe atom as Si atoms incorporate into kamacite. The studies performed are suggestive of the following. In the quartz- and feldspar- containing specimens as the result of the shock wave action there appear Fe2+ ions in the high-spin state, and, also, iron atoms in paramagnetic metallic state. All the specimens, with the exception of the olivine-containing mixture, exhibit the presence of silicon in the metallic phase. Under the action of a shock wave a local inhomogeneity increases in the neighbourhood of iron atoms in olivine whereas in enstatite there appears a local environment characteristic of the amorphous state. Apparently, for the enstatite specimen the documented amorphous state of the silicate phase is related to the occurrence of a melt glass. The inhomogeneity in the ion atoms environment in shocked olivine can be explained as disordering of the initial crystal lattice under the action of a shock wave (diapletic transformation of olivine) and the presence of neogenic olivine formed at crystallization of the shocked melt.
= 0.99+ 0/01 mm/s), characteristic of Fe2+ ions in a high-spin state, and a singlet with the shift ' = 0.05+0.03 mm/s characteristic of a Fe atom in the metallic state. Besides, the distribution function p(Hn), corresponding to the partial spectrum of kamacite in the mixture, in addition to the principal maximum at Hn337 kOe, has a local maximum at Hn305 kOe (fig.2). The presence of this maximum evidences for the appearance of a silicon atom in the closest neighbourhood of the Fe atom as Si atoms incorporate into kamacite. The studies performed are suggestive of the following. In the quartz- and feldspar- containing specimens as the result of the shock wave action there appear Fe2+ ions in the high-spin state, and, also, iron atoms in paramagnetic metallic state. All the specimens, with the exception of the olivine-containing mixture, exhibit the presence of silicon in the metallic phase. Under the action of a shock wave a local inhomogeneity increases in the neighbourhood of iron atoms in olivine whereas in enstatite there appears a local environment characteristic of the amorphous state. Apparently, for the enstatite specimen the documented amorphous state of the silicate phase is related to the occurrence of a melt glass. The inhomogeneity in the ion atoms environment in shocked olivine can be explained as disordering of the initial crystal lattice under the action of a shock wave (diapletic transformation of olivine) and the presence of neogenic olivine formed at crystallization of the shocked melt.
The obtained results directly indicate the proceeding of the impact-induced redox reaction in the silicate-melt system leading to the oxidation of iron atoms to divalent state and to the reduction of silicon atoms by the scheme
2Femetal+Si4+silicate 2Fe2+silicate+Simetal
58
Previous
Contents
Next