Crystal growth, structure and physical properties of crystals
# Chistyakova1 N.I., Rusakov V.S.2, Kozerenko S.V.2, Fadeev V.V.2, Kolpakova N.N.2 Mossbauer studies of the formation of mackinawite and tochilinite.
key words [mackinawite tochilinite Mossbauer spectroscopy]1-Lomonosov State University, Moscow, Russia; 2-Vernadsky Institute of Geochemistry and Analytical Chemistry, Moscow
The interest of investigations in respect of mackinawite and tochilinite formation is determined by the abundance of these minerals in terrestrial and extraterrestrial conditions[1]. It is known that mackinawite is tetragonal iron monosulfide. Tochilinite is an iron hydroxide -sulfide with mixed layer crystal structure consisting of alternating mackinawite tetrahedral FeS nets and (Fe,Mg)(OH)2 brucitelike octahedral layers. The aim of this study is to investigate the synthetic mackinawite and tochilinite by use of Mossbauer spectroscopy for the determination of their crystal structure and possible conditions of formation.
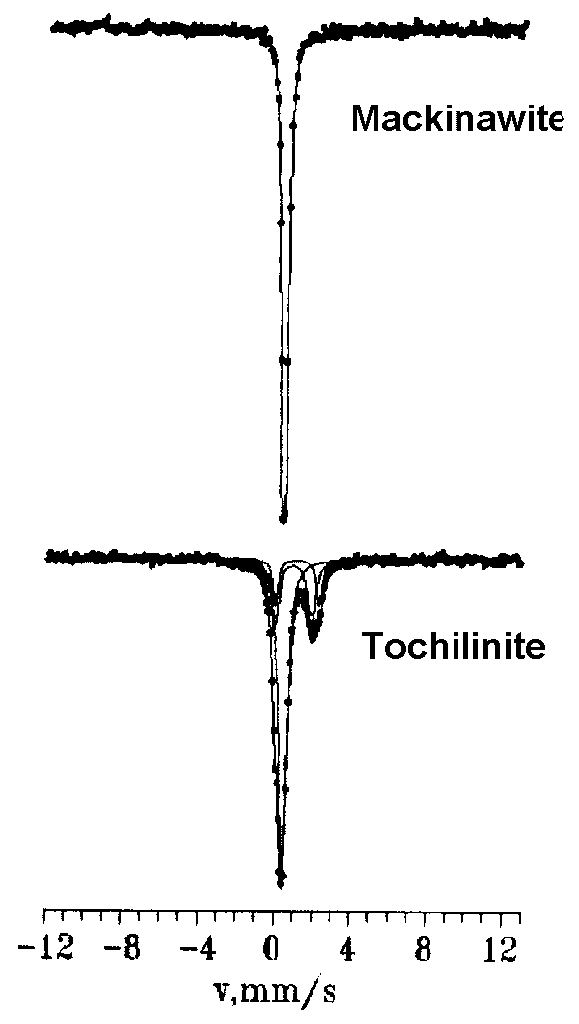
Fig.1. Mossbauer spectra of the mackinawite and tochilinite
We obtained and interpreted the Mossbauer spectroscopy of the tochilinite and mackinawite synthesized within a broad range of parameters: synthesis temperature, pH values synthesis duration, different Fe and Mg contents of the initial charge. It was shown that the mackinawite spectra obtained in the case of samples synthesized at the temperature of 160oC in the alkaline medium should be interpreted as narrow quadruple doublets (sometimes sextets corresponding to magnetite were identified) being a result of the single position of Fe ions in the mackinawite crystal structure (Fig.1). The shift of the Mossbauer line - is varied for different samples in the range of 0.37-0.39 mm/s as well as the quadruple shift of spectra components 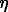 was found in the interval of 0.05-0.07 mm/s being possibly a result of different nonstoichiometric relations of synthesized samples. The values of
was found in the interval of 0.05-0.07 mm/s being possibly a result of different nonstoichiometric relations of synthesized samples. The values of 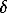 and
correspond to the hyperfine Fe2+ ion parameters in the low spin state being in the agreement with neutronographic studies.
and
correspond to the hyperfine Fe2+ ion parameters in the low spin state being in the agreement with neutronographic studies.
The Mossbauer investigations of the tochilinite were carried out. Tochilinite was synthesized in the laboratory conditions at the presence of alkaline solution (pH>8) and fixed sulfide sulfur concentration [2] at temperature range from 80 to 200oC. The Mossbauer spectra obtained are identified as the superposition of three quadruple doublets, one of them corresponding to the Fe2+ ion positions in a sulfide layer (low spin state), whereas two others belong to different Fe2+ ion positions in a brucite layer (high spin state, Fig.1).
The comparative study of hyperfine spectrum parameters of 57Fe nuclei in the mackinawite structure with that of the tochilinite sulfide layers allowed to indicate the differences in the values of
(Mossbauer line shift) and values of
(quadruple shift) in the spectra of two minerals. The increase of
for Fe2+ ions in sulfide layers of the tochilinite in relation to the mackinawite is greatly caused by the extension of elementary cell base of the mackinawite in the conjunction with the brucite cell: the increase of
is due to the distortion of the mackinawite elementary cell in the formation of the tochilinite crystal structure.
The comparative analysis of tochilinite Mossbauer spectra of samples with different Fe and Mg contents in the initial charge was performed. The dependence of relative intensity of quadruple doublets corresponding to Fe-ions in a brucite layers in relation to Mg content is consistent with the calculated relation of the number of Fe ions in a brucite layer to the total number of Fe ions in the tochilinite structure. The calculations were carried out on the assumption of the same thickness of mackinawite and brucite layers. These results indicated that mackinawite layers are in the conjunction with the equal number of brucite layers but this number presumably is not above 2.
# The work has been supported by the Russian Foundation for Basic Research
81
Besides it was found that the Mg2+ ions are present only in a brucite layer of the tochilinite crystal structure being located preferentially in a single position as the relative intensity of one of the subspectra was found to be practically invariable at variable Mg content.
References:
Novikov G.V. , and Sipavina L.V. Ge-pyroxenes: structure features, phase transitions, and the local fields on 57Fe nuclei.
key words [germanium pyroxene phase transition]
Due to the great importance for mineralogy the crystal structures of pyroxenes, their stability fields and phase transitions are the subject of intensive studies by different methods. In particular, the crystal-chemical motives and mechanisms of high pressure and high temperature phase transformations of pyroxenes are interesting because of their notorious role in the mineralogical models of the Earth's mantle. Chain germanates are good reference system for the study of specific pyroxene structures with rigid tetrahedral groups, that possess to a considerable extent the geometrically limited structures. The comparative analysis of the structure features, geometries of metal polyhedra, and the metal-oxygen bonds in pyroxenes and in relative germanates was not carried out so far. We are in a position to present some data concerning crystal chemistry of mantle related pyroxene structures.
In this work the Ca-Fe chain germanates with two distinct monoclinic structures and Fe-Mg germanates with orthorhombic structure were produced at normal pressure by solid state reaction at temperatures 900-1050oC and studied by X-ray powder diffraction and 57Fe gamma-resonance methods. One of the goal of the study was to compare both electronic states of Fe2+ and the oxygen polyhedra parameters in germanates and relative silicates with typical pyroxene structures.
To determine the structure features of germanates, their X-ray powder diffraction patterns were received by step scanning method and the parameters of partial Bragg reflexes were evaluated. The angle positions and relative intensities of 25-32 well determined reflexes could be evaluated. The unit cell parameters of germanates were determined by the least squares method.
The unit cell parameters a, b, c, and angle b of monoclinic germanates Fe(Fe(1-x)Cax)Ge2O6 were found being changed by abrupt manner at x = 0.4, indicative of the phase transition from the Ca poor- to the Ca rich phase structure, accompanied by the volume jump of 4.4 %. The space group C2/c was supported for both types of monoclinic structures by the qualitative inspection of relative intensities of Bragg reflexes, at atomic coordinates in the Ca poor phase (0<x<0.4) being constrained identical to corresponding coordinates in MgGeO3 [1], and in hedenbergite [2] in the case of the Ca reach phase (0.4 < x <1).
To determine the atomic coordinates of ions, the attendant changes in geometries of metal polyhedra M1, M2 and tetrahedra GeO4 , as well as the O3-O3-O3 chain extension angle along the Fe-Ca join, the original three step algorithm, based on the least squares principle, was used. The starting values of atomic coordinates for the Ca poor and Ca reach phases were taken from [1] and [2], respectively. At the first step only coordinates of cations were specified, the coordinates of oxygen atoms were fixed being equal to starting values. At the second step the determined at the first step coordinates of cations were fixed and just the oxygen coordinates were refined. At the third step coordinates of all ions were varied. Using these results, geometries of polyhedra and tetrahedral chains in germanates were determined.
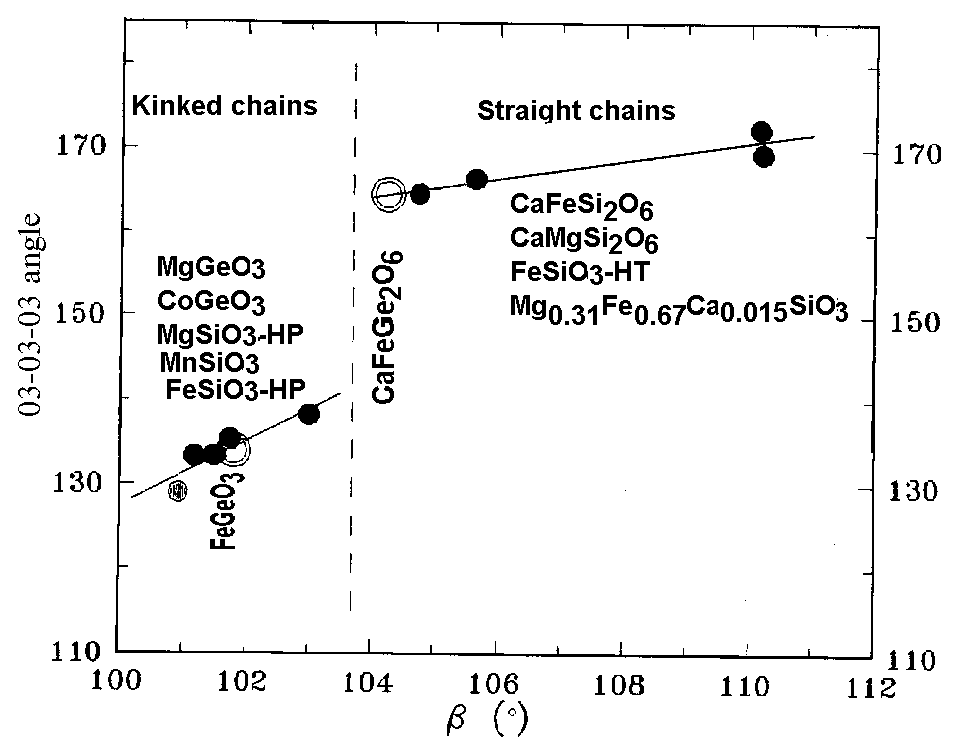
Fig 1. The plot of O3-O3-O3 chain angle against the b angle of monoclinic germanates and silicates.
Two distinct types of monoclinic structures of chain silicates and germanates with the same space group C2/c, having different kind of M2 polyhedra, can be easily distinguished, using the plot of the O3-O3-O3 angle against the monoclinic b angle (Fig. 1). Structurally, the Ca poor germanate phase (b <103.5 deg.) definitely belongs to the first group of structures (C2/c-I) with kinked chains of tetrahedra. In this structure the M2 cations are surrounded by 6 oxygen ions, which form highly distorted octahedron. This group includes the Mg-[1], Co-[3] and Mn-[4] germanates and high pressure Fe-[5] and Mg-[6] pyroxenes, which could not be retained at normal pressure. The Ca reach phase (b >103.5 deg.) is to be referred to the structures (C2/c-II) with the straight tetrahedral chains, ions at the M2 sites are surrounded by 8 oxygen ions. This group consists of Ca bearing pyroxenes
82
diopside and hedenbergite [2] and of nonquenchable high temperature form of ferrosilite FeSiO3 [7]. In all these chain structures the oxygen ions at M1 cations form the regular octahedra. The Fe2+ ions in chain silicates and germanates can be arranged both in M1 and M2 crystallographic positions.
It is worth to mention, that the structure and phase relations in the Ca bearing and Ca free pyroxenes are different in some important features just because of Ca cation size. In chain germanates, as it was determined in this work, the Ca ions are distributed in M2 polyhedra, whose 'mean' size and geometries are changing with the Ca content by abrupt manner at the phase transition C2/c-I - C2/c-II. At the same time, in these two structures, both the size and the shape of regular M1 octahedra are very close.
To understand the possible role of Fe-O bonds in M1 and M2 sites at the phase transition, found at x = 0.4, it was very interesting to trace the attendant changes in the electronic state of Fe2+ ions and compare them with the data on pyroxenes. For this the 57Fe 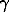 -resonance was used. Ordinary the Fe2+ ions at two geometrically distinct polyhedra M1 and M2 in chain structures give two-doublet 57Fe spectra, and local fields on 57Fe nuclei at these sites can be determined accurately. The room temperature 57Fe spectra of Fe-Ca germanates with both C2/c(I) and C2/(II) structures consisted, at first sight, of two 'doublets' (Fig.2, x(Ca)=0.8, top), which do not reflect any dramatic changes in the hyperfine parameters of these Fe2+ 'doublets' at this phase transition. The liquid nitrogen spectra of C2/c(II) phase give again 'two doublets' (Fig.2, x(Ca)=0.8 bottom), which consisted, however, of more broadened components, having unrealistic profiles. What is more important, the relative intensities of these 'doublets' differ at these two temperatures very much, proving, that, at least, these low temperature spectra do not consist, in real, of two doublets.
-resonance was used. Ordinary the Fe2+ ions at two geometrically distinct polyhedra M1 and M2 in chain structures give two-doublet 57Fe spectra, and local fields on 57Fe nuclei at these sites can be determined accurately. The room temperature 57Fe spectra of Fe-Ca germanates with both C2/c(I) and C2/(II) structures consisted, at first sight, of two 'doublets' (Fig.2, x(Ca)=0.8, top), which do not reflect any dramatic changes in the hyperfine parameters of these Fe2+ 'doublets' at this phase transition. The liquid nitrogen spectra of C2/c(II) phase give again 'two doublets' (Fig.2, x(Ca)=0.8 bottom), which consisted, however, of more broadened components, having unrealistic profiles. What is more important, the relative intensities of these 'doublets' differ at these two temperatures very much, proving, that, at least, these low temperature spectra do not consist, in real, of two doublets.
To improve the effective spectroscopic resolution in spectra (up to 2-3 times) at a cost of signal-to-nose ratio, we used the original mathematical N-procedure [8], but no any additional partial spectral components could be distinguished in such N-spectra neither at room temperature, nor at 80K (Fig.2, T=300K, 80K, x(Ca)=0.8). The detailed numerical analysis has shown that the spectra of the C2/c(I) phase (x < 0.4) in paramagnetic state consisted of two doublets with narrow components, having the profile, close to Lorenzian. Spectra of C2/c-II phase are more complex and at temperatures 120-220K two additional components appear and three doublets could be definitely distinguished in the spectra. The example of the three-doublet approximation of one of the N-spectra (at 160K) is shown in Fig.2, T=160K, x(Ca)=0.8).
The hyperfine (HF) data for Fe(Fe0.2Ca0.8)Ge2O6 at temperatures 120-220K are presented as a plot of the quadrupole splitting (QS) against isomer shift (IS), which permits one to determine three different types of electronic state of Fe2+ ions in the C2/c-II phase. Assigning these three doublets to crystallographic positions, we are to propose three (Fig.3) different electronic states of Fe2+ in two crystallographic positions M1 and M2.
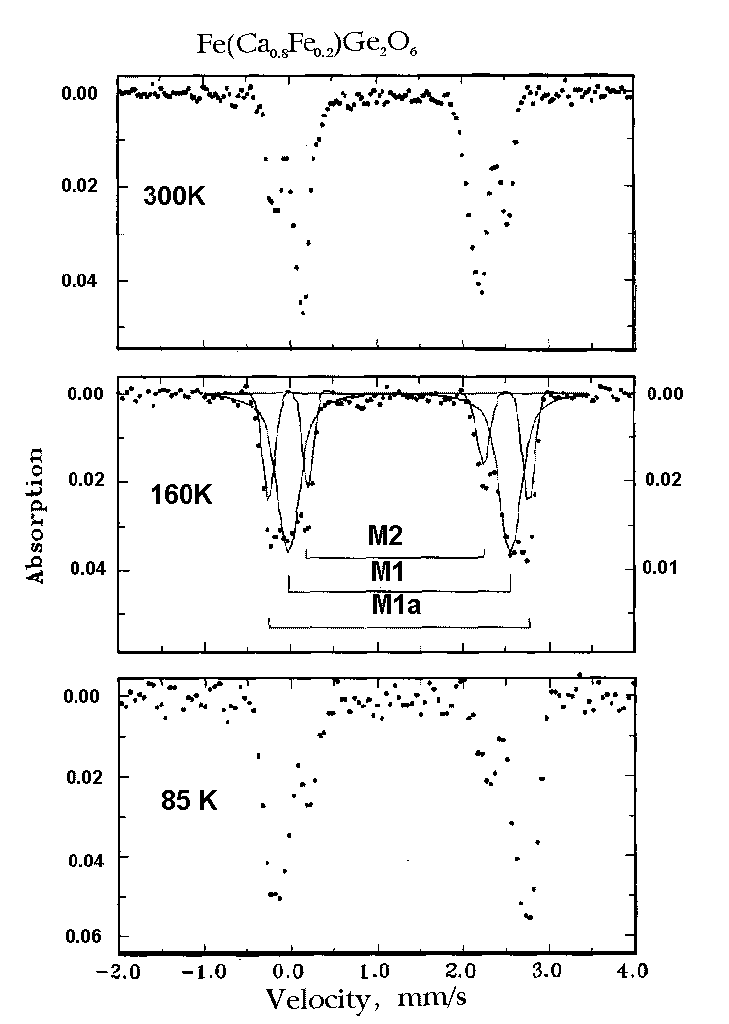
Fig. 2. The N- spectra of Fe(Fe0.2Ca0.8)Ge2O6 at 300K (top), 160K (middle), and 85K (bottom).
Fig. 3. Plot IS against QS for three distinct electronic states of Fe 2+ at 120 - 220K, corresponding to M1, M1a, and M2 doublets in spectra of Fe(Fe0.2Ca0.8)Ge2O6 .
Assignment of these doublets to M1 and M2 positions is shown in Fig.2 (middle). Iron in M1 sites gives two doublets: M1 and M1a, M2 sites give one doublet. There are the following arguments for this conclusion. 1.Most of Fe2+ ions occupy the M1 sites, and the most intensive doublet (M1) is to be referred to this octahedra. 2. The Fe2+ ions in the very distorted M2 sites always demonstrate small value and very week temperature dependence of QS. So, the doublet with smallest QS belongs to the
83
iron ions in M2 sites. 3. The high value of QS as well as their temperature dependence permit us to refer the M1a doublet to M1 sites.
This assignment of doublets is supported by comparison of hyperfine parameters QS and IS of C2/c-II phase with the HF parameters of well known silicates: ferrosilite [9] and hedenbergite [10]. Most of Fe2+ ions in germanate Fe(Fe0.2Ca0.8)Ge2O6 have the HF parameters, close to those in hedenbergite (M1 doublet, Fig 2). According to our results geometry of M1 octahedra in the Ca rich phase is close to that in hedenbergite. Other part of Fe2+ in the M1 sites (doublet M1a) has QS and IS, which are close to the parameters of Fe at M1 sites in ferrosilite [9]. It means, that the M1 polyhedra of two sorts are to exist in the structure C2/c-II.
Fe2+ ions at the M2 sites (doublet M2, Fig. 2) have HF parameters, typical of M2 positions in all chain structures, including their most remarkable feature - QS is changing with T very slightly.
In some conclusion, we found three contrast electronic states of Fe2+ in the Ca rich Ca-Fe germanates, what is characteristic just for the C2/c-II structure, in contrast to the C2/c-I structure of Ca poor phase. The point is, that in this two structures just M2 sites differ in their geometries, but the Fe ions at another, M1 site, have different electronic structure. For all we know, this phenomenon was not published in literature. We guess, that in some silicates this phenomenon can be also found.
The results of the study of Mg-Fe chain germanates have shown, that at T about 1000oC and at normal pressure Mg ions stabilize the orthorhombic structure with the Pbca space group. Just at high Fe content (more than 70%) the monoclinic C2/c structure can be synthesized and retained by quenching. Depending on the synthesis temperature the end member, FeGeO3 , can be produced and saved by quenching both with monoclinic and orthorhombic structures, however, the really perfect phase of orthorhombic FeGeO3, apparently, can not be synthesized easily at normal pressure [11].
In paramagnetic state Mg-Fe orthogermanates give the spectra, which can be, at first sight, approximated by two doublets, as relative pyroxenes. However, the components in spectra of orthorhombic germanates are broadened and, what is more important, their profile is far away from the Lorenzian one both at room temperature and at 80K. There is one more, apparently the most trouble fact - the relative intensities of 'two doubles' are definitely different at the room- and at the liquid nitrogen temperatures, if one fits the two-doublet model to the experimental spectra of Mg-Fe germanates. So, we can not be sure, if the spectra consist just of two doublets. If, nevertheless, we assign two formally distinguished doublets to M1 and to M2 crystallographic positions, their room temperature HF parameters can be compared with the corresponding data for the Fe-Mg orthopyroxenes.
The isomer shifts of these distinguishable 'two doublets' in Fe-Mg germanates are practically equal, in contrast with the Fe-Mg pyroxenes - the IS of Fe2+ in M1 site is definitely higher relative to the IS of M2 site in pyroxenes. It is the most remarkable difference in HF-parameters of orthorhombic Mg-Fe germanates and silicates.
The detailed numerical analysis of 57Fe spectra of Fe-Mg chain germanates shows some specific features. We are careful in conclusions, because these features resemble the features, which accompany the phenomenon, found for paramagnetic Ca-Fe germanates, presented in this paper. Additional investigations are needed without fail to complete the interpretation of data on these orthorhombic Fe-Mg germanates.
References:
Karaseva O.N., Lakshtanov L.Z., Ivanova L.I. Complexation of strontium on the surface of hematite.
key words [strontium adsorption hematite]
Adsorption on the surfaces of minerals is one of the most important factors which govern the composition of hydrothermal solutions. The account taken of the adsorption properties is also important for solving the problems
84
of the enviromental control, in particular, for estimation and forecast of immobilization of radioactive waste.
Among the most dangerous radioactive materials is 90Sr isotope forming in nuclear reactions of uranium fission. In our experiments we used as a sorbent iron oxide which is one of the most abundant components of the continental crust.
The main goals of the work are as follows: the determination of the stoichiometry of surface Sr complexes and the creation of a thermodynamic model of the heterophase system involving the surface of hematite and the solution of the background electrolyte, containing Sr. The adsorption equilibria are herewith considered as the complexation reactions of strontium with hydroxyl groups on the hematite surface.
Experimental methods and results. The adsorption of strontium at the background of 0.1 M NaCl solution was studied by the method of potentiometric titration. All the runs were carried out in a hermetic thermostated cell at T=25oC.
Each run on Sr adsorption was preceded by a study of the acidobasic properties of hematite the account of which is necessary for the creation of a thermodynamic model of the H+-hematite surface - Sr2+ system. In the course of each titration of hematite the total amount was determined of hydroxyl groups º FeOH which form on the mineral surface at the contact with the water solution and which participate in the protonation and deprotonation reactions and in the processes of complexation with metal ions. The concentration of º FeOH was estimated from the experimental data obtained in the region of complete protonation of the hemotite surface in the range 2.7<pH<3.0 to be 2.0+0.05 mM. The determination of the constants of the acido-basic equilibria was carried out using the model of the constant capacitance of the double electric layer (Table 1).
The adsorption of Sr was studied in the pH range from 6 to 11 for the values of the Sr concentration in the solution 0.2, 1.0, 2.0 mM.
It was found that the adsorption of Sr with the concentration of 0.2 mM begins at pH 7.2 and reaches 95% at pH 10. As the concentration of Sr grows, the degree of its adsorption decreases.
The principle equation of the complexation reaction on the surface of hematite, involving H+, Sr2+, and º FeOH may be represented as
pH+ +q(º
FeOH) + r Sr2+ ¬
 Hp (º
FeOH)qSrr(p+2r)
Hp (º
FeOH)qSrr(p+2r) 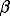 sp,q,r
sp,q,r
The quantities
sp,q,r are conventional constants; in order to obtain the corresponding thermodynamic constants one has to introduce the correction for the electrostatic potential of the charged surface:
sp,q,r (int) =
sp,q,r ×
e ((p+2r)Fy
/RT)
where y is the electrostatic surface potential.
The calculation of the H+- º FeOH - Sr2+ model consisted of testing the possible combinations of the surface complexes of strontium. Best of all the experimental data fit the model including the formation of three surface complexes. Given on the diagram (fig.1) are the experimental data on the adsorption of strontium and those calculated using the constant capacitance model. The equations of the complexation reactions and the corresponding thermodynamic constants are listed in the table.
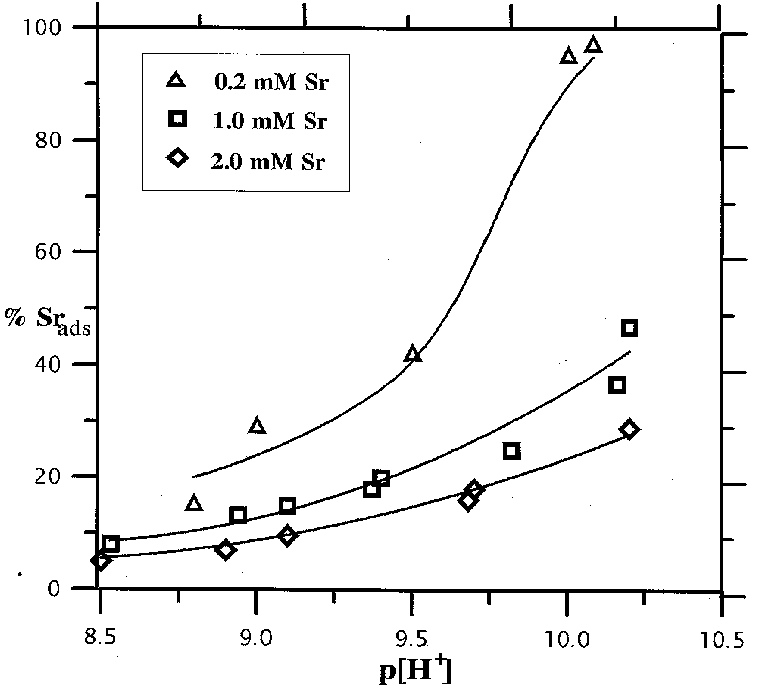
Fig.1. Sr absorption at the surface of hematite (surface area - 6 m2/g, concentration of suspension - 70 g/l. Total concentration of absorption sites - 2.0 mM). The lines represent the proposed model.
Table 1. Surface complexation reaction model (+3s ) in the H+-hematite (º FeOH)-Sr2+ system
|
Surface equilibria |
log |
+3s |
|
º FeOH + H+ ¬ ¾ ® º FeOH2+ |
7.31 |
0.03 |
|
º FeOH ¬ ¾ ® º FeO- + H+ |
-9.72 |
0.01 |
|
º FeOH + Sr2+ ¬ ¾ ® º FeOHSr2+ |
1.85 |
0.01 |
|
º FeOH + Sr2+ ¬ ¾ ® º FeOSr+ + H+ |
-8.07 |
0.06 |
|
º FeOH + Sr2+ + H2O ¬ ¾ ® º FeOSrOH + 2H+ |
-18.04 |
0.07 |
|
specific capacitance 2.9 F/m2 |
85
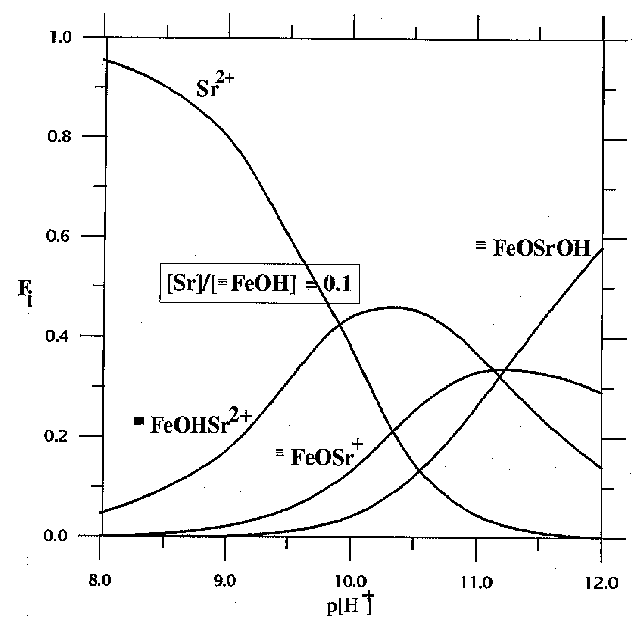
Fig.2. Distribution diagram for Sr, Fj denotes the fraction of the total Sr concentration in each species
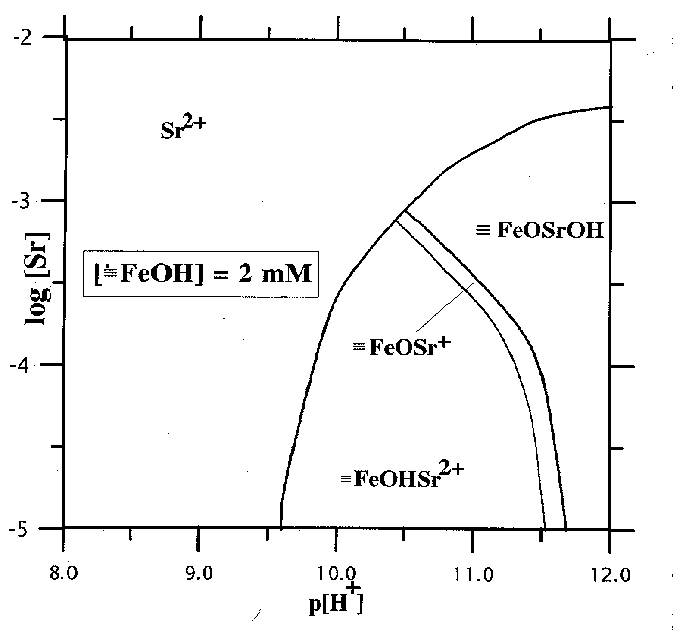
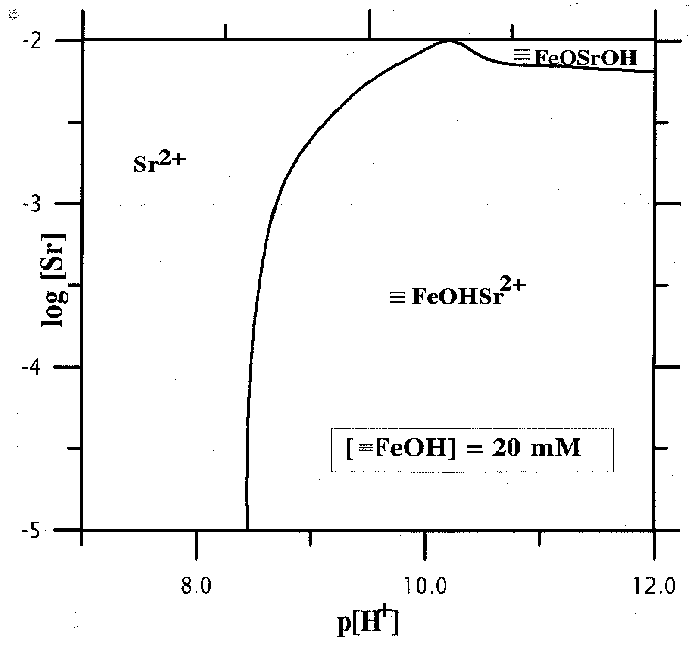
Fig.3. Predominance area diagrams for Sr species.
Conclusions.
1. The adsorption of strontium occurs preferentially in strongly alkaline region growing with the growth of pH from 7 to 11 and the decrease of the initial concentration of Sr in the solution (fig.1).2.The complexation reactions of Sr with the surface OH-groups give rise to surface º FeOHSr2+ , º FeOSr+, º FeOSrOH complexes. The diagrams of the distribution of Sr particles in the solution and on the surface for the given concentration of Sr as a function of pH of the suspension are shown in fig.2. It is noteworthy that at low [Sr2+] practically all the adsorption is governed by the formation of two-charge complex on the mineral surface. Provided that the concentration of Sr is strong, the neutral complex º FeOSrOH, forming at pH of the suspension in excess of 10, prevails in the system.
3. The sorption of Sr is strongly affected by the concentration of hydroxyl groups on the surface (fig.3). As it grows by 10 times, pH of the Sr adsorption decreases by greater than 1.
4. So, the adsorption of Sr not characteristic of ground water with the close-to-neutral pH, can be determinant for radioactive waste disposal under the conditions of high pH of the medium.
Dadze T.P., Fediouchtchenko S.V., Koschug D.G., Shvarov Yu.V. Li promoted substitution of Al for Si in quartz.
key words [quartz lithium aluminium synthesis]
It is well known that heterovalent substitution of M3+ cations for Si in quartz is accompanied by entering of M+ charge compensating ions. Al3+ is the very common impurity ion. M+ ions such as H+, Li+, Na+ and K+ are located
86
in structural channels running along z-axis [1]. Presence of H+ was revealed by infrared spectroscopy whereas presence of Li+, Na+, and K+ is not proved definitely. Up to date there is no certain experimental evidence for the role of different charge compensating ions on the substitution of Al for Si. At the same time structural Al impurity is often used as an index of physico-chemical parameters of rock-forming media, P-T parameters of metamorphism, etc.
The main goal of this work was to find out the influence of different M+ ions on Al substitution for Si in quartz upon hydrothermal recrystallization.
Natural rock crystal was crushed, sieved, and size fraction of 0.1-0.4 mm separated for the recrystallization experiments. Titanium autoclaves and platinum, gold or silver ampoules (depending on the composition of hydrothermal solution) have been used. Experiments were carried out mainly at 400oC and 0.05 GPa. Usual duration of each experiment was 14 days. Chemical composition of hydrothermal solutions was controlled by HCl, NaOH and LiCl. It varied from acid to basic solutions: 0.1, 0.01, 0.001 mole HCl, H2O, 0.002, 0.02, 0.2 mole NaOH. Concentration of LiCl was 0.0025, 0.005 and 0.01 mole. The weight of quartz was 0.1g, H2O volume � 3 to 11 ml. Feldspar (0.1 g) was used as a source for Al, Na, and K in the fluid. The composition of feldspar (Ab60Or40) was determined with electron microprobe analysis.
The equilibrium compositions of the experimental systems at elevated temperatures have been calculated with the software package HCl [2] using the Gibbs free energy minimization technique.
Al concentration in quartz was measured with electron paramagnetic resonance (EPR) spectroscopy because it refers to structural impurities excluding Al possibly located in mineral inclusions. EPR measurements were carried out with Varian E-line spectrometer at 77 K. The microwave power was held at 10 mW, modulation amplitude was 10 Oe. Intensity of the line at geff=1.993 being well resolved was selected for the estimation of Al content. Before EPR measurements quartz samples were
-irradiated (60Co source) with an analytical dose of 106 Gy.
Three series of experiments have been done: 1) recrystallization without feldspar and LiCl; 2) recrystallization with the presence of feldspar without LiCl; and 3) recrystallization with the presence of feldspar and LiCl. These series pointed out to reveal the influence of H+ 1), Na+ and K+ 2), and Li+ 3) on heterovalent substitution of Al for Si. The initial concentration of Al in quartz before recrystallization was about 30 ppm. It decreased upon the recrystallization in neutral and acid solutions for the first and second series (fig. 1). In these cases Al content is controlled by the surface charge of quartz, i.e., by the concentration of dissociated surface Si-OH groups [3]. There was no visible influence of H+, Na+, and K+ on the concentration of Al in the recrystallized quartz. Most probably that charged surface sites and neutral Al species of the fluid are involved in the Al incorporation process by growing quartz.
The third type of experiments shows that influence of LiCl on the Al concentration in quartz is completely different (Fig.). Content of Al increases after recrystallization by a factor of 1.5 to 3 even in acid solutions. Unfortunately, for the moment, dominating fluid Li species cannot be obtained by means of equilibrium thermodynamic calculations. Nevertheless, these results confirm the prevailing role of Li in the substitution of Al for Si in quartz which is concordant with the results of investigation of natural quartz [4].
Al concentration in hydrothermally recrystallized quartz
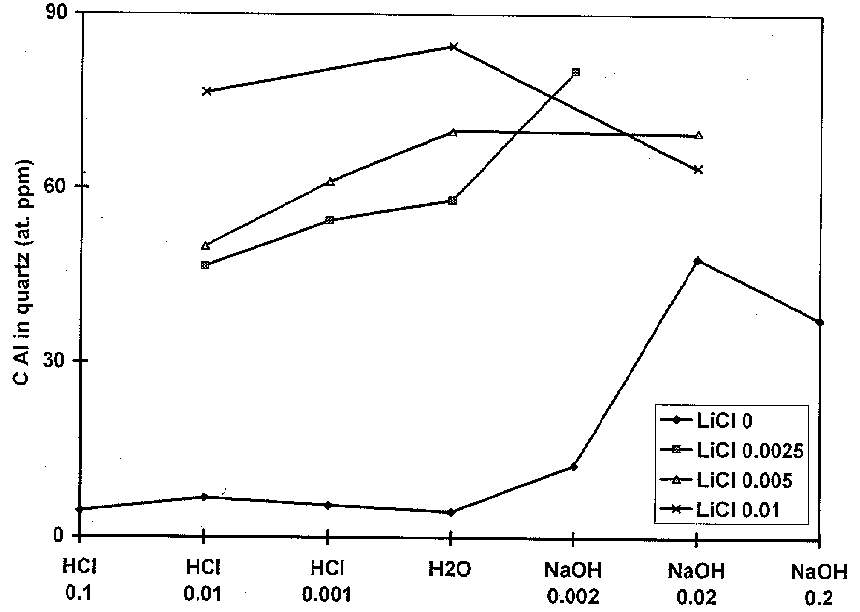
Composition of hydrothermal solutions(M)
References:
Bulbak T.A., Shvedenkov G.Yu., Lepezin G.G. Experimental data on the substitution of D2O and CO2 for H2O in cordierite channels.
key words [Mg-cordierite fluid metamorphism CO2 H2O in cordierite channels]
Cordierite whose structure carries cavities and channels - the sites of localization of volatile components is quite promising from the viewpoint of reconstruction of the fluid regime of natural hydrothermal and metamorphogeneous mineral formation. Due to their rather low diffusion coefficients this mineral can be recommended as a 'sampler' of a mineral-forming fluid.
In this work we have reproduced experimentally the process of adaptation of the channels contents to the surrounding fluid. Synthetic magnesian cordierite (indialite), preliminary saturated with water at T=640oC and P=250MPa for 88 h, was immersed into the D2O or CO2
87
medium at T=700oC and P=200MPa (for 113-137h). The runs were performed on an apparatus HPA-1000. CO2 was generated by decomposition of silver oxalate. In order to avoid the liberation of volatiles by the mineral the 'quenching' was performed by the isochore of water at temperatures down to 400oC and below 400oC - at a pressure of 80-100 MPa. The containers were gold capsules electrowelded under cooling with liquid nitrogen.
The IR-spectroscopy and quantitative chromatography data show that at the background of the decrease of the total water content by 0.2 wt% there occurs the substitution of D2O for water in the channels by at least 50%. In the CO2 medium water is expelled from its positions by approximately 64%. The specific features of distribution of the cordierite particles sizes before and after the intercalation indicate the absence of the recrystallization processes.
Our studies suggest the following most important conclusions.
H2O molecules of two types have been documented in synthetic magnesian cordierites. This evidences for the fact that their orientation is affected not only by cations of alkaline elements, but other factors as well including, possibly, the processes of structure ordering.
Cordierite acts as a peculiar sensor. In particular, if the fluid consists of H2O and CO2 , then water will be preferential in entering the channels.
The treatment of H2O-saturated cordierite with heavy water leads to the complete displacement of type I H2O molecules and a noticeable substitution of type II molecules. The process is rapid enough and is, possibly, realized by the self-diffusion mechanism.
So, we were the first to establish experimentally the fact of exchanging guest molecules between channels of the cordierite structure and the surrounding fluid.
Bulbak T.A., Shvedenkov G.Yu. , Lepezin G.G. Behavior of hydrocarbons in synthetic cordierites under pressure.
key words [Mg-cordierite sensor hydrocarbons fluid metamorphism]
Joint Institute of Geology, Geophysics and Mineralogy SB RAS Novosibirsk Russia
Cordierite is one of the not many minerals capable, due to its structural features, of trapping volatile components which were in the former time constituents of a mineral-forming fluid. In natural specimens water and carbon dioxide dominate quantitatively. The concentration of other components (hydrocarbons, Ar, N2, He) is insignificant. As suggested by the mass-spectroscopy data [1] the concentration of hydrocarbons is 1.5 g/kg in the samples from a granulite facies. The IR-spectra of the unheated samples demonstrate strong lines in the region 3000-2800 cm-1 responsible for the occurrence of alkanes [2,3]. The question is: to what extent do the contents of the channels of the mineral under study correspond to the fluid composition at high T- and P-parameters? This work has been devoted to the experimental verification of this correspondence.
The runs were conducted as follows: synthetic magnesian cordierite (indialite) was placed into a reducing medium and kept at a temperature of 700oC and pressures 200 and 1000 MPa. The forms of occurrence of volatiles were defined by the IR-spectroscopy, and the character of release and the amount of volatiles were derived from the chromatography results.
The experiments were run on the apparatus HPA-1000 [4] under moderate pressures (for about 130 h) and piston-cylinder type apparatus [5] under high pressures (for about 8 h). The initial material was synthesized from 2MgO-2Al2O3-5SiO2 at T=1400oC and P=0.1 MPa , the duration of the synthesis was 1 h.
The mineral powder (mean particle size ~
30  m) was then placed into preevacuated gold capsules. The capsules were filled with gases of methane-butane series (national standards of hydrocarbons), hermetically pinched and electrowelded (under cooling with liquid nitrogen). The comparison of the IR-spectra of cordierites kept under moderate-and-high-pressure suggest a number of conclusions. Despite the measures taken to ensure the sterility of the capsule filling, all the spectra demonstrate the absorption of trace amounts of water (types I and II). This may be related with the formation of H2O due to oxygen from the mineral carcass and hydrogen generation by the destruction of hydrocarbons. C-H vibrations in the methane molecules (3018 cm-1) were found only under moderate pressures in 'dry' and methane-water environment. Practically all the spectra demonstrate absorption lines of the benzene ring (1605, 1570,1470 cm-1). The spectra of cordierites kept under moderate pressures in C2H6-C4H10 do not virtually differ. The samples saturated under high pressures demonstrated 2925, 2860 cm-1 lines, corresponding to stretching C-H vibrations in CH2- CH3 -fragments.
m) was then placed into preevacuated gold capsules. The capsules were filled with gases of methane-butane series (national standards of hydrocarbons), hermetically pinched and electrowelded (under cooling with liquid nitrogen). The comparison of the IR-spectra of cordierites kept under moderate-and-high-pressure suggest a number of conclusions. Despite the measures taken to ensure the sterility of the capsule filling, all the spectra demonstrate the absorption of trace amounts of water (types I and II). This may be related with the formation of H2O due to oxygen from the mineral carcass and hydrogen generation by the destruction of hydrocarbons. C-H vibrations in the methane molecules (3018 cm-1) were found only under moderate pressures in 'dry' and methane-water environment. Practically all the spectra demonstrate absorption lines of the benzene ring (1605, 1570,1470 cm-1). The spectra of cordierites kept under moderate pressures in C2H6-C4H10 do not virtually differ. The samples saturated under high pressures demonstrated 2925, 2860 cm-1 lines, corresponding to stretching C-H vibrations in CH2- CH3 -fragments.
With the ethanol filler in the capsule (P=1000 MPa) there appear additional features in the spectrum, i.e. two 1780 and 1680 cm-1 lines assigned to the C=O vibrations in aromatic carbonic acids or their aldehydes, and the absorption of the benzene ring with the frequency of 1555 cm-1, not found under other conditions.
The maximum concentrations of hydrocarbons obtained by quantitative chromatography (in mg/kg sample) are:
CH4=78.3; C2H6=134; C2H4 + C2H6 = 26; C3H8=28, C4H10=32; C5H12=23; C6H14=7. The total content of hydrocarbons extracted from the sample varies as follows (the composition of the gas before the run, mg/kg sample):
P=200 MPa CH4=21.9; C2H6=130.9; C3H8=120.4, C4H10=54.4; P=1000 MPa CH4=105.4; C2H6=126; C3H8=323.3, C4H10=117.7.
As seen, the effect of pressure shows up in the absolute increase in the concentration of the 'organics' in the mineral, excluding the case of ethane in the capsule.
So, the incorporation of hydrocarbons into the structure of cordierites has been experimentally proved. Herewith the forms of their occurrence and precipitation do not coincide, therefore they reflect the fluid regime not directly (like in the case of H2O-CO2), but indirectly.
88
References:
#
Litvin Yu.A., Chichagov A.V., Bondarenko G.V. Polymorphism of Na2Mg2Si2O7 at high pressures: X-ray diffraction and IR-spectroscopic characteristics of a plausible mineral of the mantle.key words [Na-Mg silicates Na2Mg2Si2O7 high pressure polymorphism]
Na-Mg silicates, i.e. Na2MgSiO4, Na2Mg2Si2O7, Na4Mg2Si13O10, Na2Mg2Si6O15, and Na2Mg5Si12O30 have arosed considerable interest as plausible minerals of the Earth's mantle after it was found in the high-pressure experiments (Litvin, Gasparik, 1995; Gasparik, Litvin, 1997) that forsterite reacts with jadeite under pressures in excess of 4.5 GPa yielding pyrope, jadeite-enstatitic clinopyroxene and the little known compound Na2Mg2Si2O7. In nature only Na-Mg-silicate Na2Mg5Si12O30 is encountered as a basic component of solid solutions of the meteorite mineral - roederite (the second component is the potassium analogue). Na2Mg2Si2O7 was then obtained in the reaction of enstatite with nepheline (Gasparik, Litvin, 1997). The PT-curve of Na2Mg2Si2O7 melting was studied in the range from 1 atm to 22 GPa (Litvin et al, 1996; Gasparik, Litvin, 1997). The compound was found to be most fusible as compared with the known mantle minerals.
The experimental studies performed at high pressures to 7 GPa and temperatures to 1500oC have shown that all the afore cited Na-Mg-silicates, with the exception of Na2Mg2Si2O7 , disproportionate under the effect of pressure to yield simpler compounds (Na2SiO3, MgO, MgSiO3, SiO2) within 3-6 GPa.
The compound Na2Mg2Si2O7 undergoes a polymorphic transition at pressures in excess of 3.6 GPa to a new yet unknown modification -
-phase. The high pressure phase exibits the ability of being metastable at 1 atm. The new polymorphic species Na2Mg2Si2O7 has been identified from the X-ray diffraction data (Table 1) and from the IR-spectroscopy data (Table 2).
Table 1. X-ray diffraction data.
|
|
|
|
|
||||
|
d |
I/Io |
d |
I/Io |
d |
I/Io |
d |
I/Io |
|
4.3412 |
42 |
2.268 |
21 |
||||
|
4.239 |
100 |
2.1358 |
24 |
||||
|
2.767 |
26 |
1.9910 |
25 |
||||
|
2.625 |
85 |
1.9786 |
24 |
||||
|
2.580 |
53 |
2.5587 |
61 |
1.505 |
10 |
1.9473 |
15 |
|
1.477 |
13 |
||||||
|
2.510 |
27 |
||||||
|
2.5007 |
100 |
1.4423 |
38 |
||||
|
2.456 |
31 |
||||||
Note: X-ray diffraction characteristics of the phases were obtained by digital recording of the reflections in a discrete mode in an automatic diffractometer HZG 4/PC/AT, 35 kV and 30 mA, CoK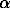 (in the case of the
-phase) and CuK
(in the case of the
-phase) radiations,
-filter, external standard Si, a=5.4305�.
(in the case of the
-phase) and CuK
(in the case of the
-phase) radiations,
-filter, external standard Si, a=5.4305�.
Table 2. IR-spectroscopy data (the absorption bands positions are given in cm-1).
|
|
|
|
|
|
294 |
829 |
||
|
358 |
863 |
||
|
424 |
899 |
||
|
469 |
912 |
||
|
477 |
988 |
||
|
507 |
993 |
||
|
545 |
1034 |
||
|
612 |
612 |
1442 |
Note: The infrared absorption spectra were obtained in a spectrophotometer Perkin Elmer mod 983. Compacted tables of KBr homogeneously intermixed with fine-disperse powders of
- and
-phase samples were used. The spectra were recorded in the frequency range from 250 to 1600 cm-1 with the resolution
3 cm-1.
As a result low- and high-pressure phases of Na2Mg2Si2O7 were unambiguously identified as individual crystalline substances. The phase compositions were controlled by micro- X ray spectral analysis (spectrometer Camebax). The standardised X-ray diffraction and IR-spectroscopic characteristics of the new high-pressure polymorphous modification
-Na2Mg2Si2O7 were first obtained.
89
The runs at high pressures and high temperatures were carried out using 'an anvil-with-hole' apparatus .
Poltavets Yu.A. Ti and Fe3+/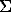 Fe ratio in garnets as measure of a relative depth of the garnet-bearing rock formation in the lower continental crust and in the upper mantle.
Fe ratio in garnets as measure of a relative depth of the garnet-bearing rock formation in the lower continental crust and in the upper mantle.
key words [garnet titanium ferric-ferrous relation]Institute of Geology and Geochemistry UB RAS Ekaterinburg
The composition of minerals yields the main information on the physicochemical conditions of the minerals formation. In this respect such minerals as olivines, ortho- and clinopyroxenes, chromspinelides are studied at greatest length whereas spinelides of the magnetite series and garnets are less studied [1-6]. In this work an attempt has been made to use the published experimental data on the composition of garnets to interpret the specific features of the behaviour of certain garnet components, exhibiting clear dependencies on the PT-conditions of their formation in garnet-bearing rocks of various deep facies.
Fig.1. Geochemical features of the garnet composition in the upper mantle rocks. The diagram is plotted using the data of Green D.H., Ringwood A.E. et al [3.] 1-gabbroid or pyroxene granulites (plagioclase + pyroxene + olivine + spinel); 2-garnet granulites (garnet +pyroxene + plagioclase) ; 3-eclogites (garnet +pyroxene + quartz); 4-garnet peridotite (forsterite + alumina -depleted pyroxene +garnet) from oceanic regions; 5 - garnet+peridotite from continental regions; 6 - subliquidus associations region; garnet - co-existing with liquid; 7-9 isopleths of pyrope/almandine ratios, titanium (8) and the degree of oxidation of iron (Fe3+/
Fe ) in garnets; 10 - geothermal gradients of oceanic (o) and continental (c) regions.
The statistic analysis of the garnet composition data [1-3, 5,6] has revealed for some elements a number of interesting features; in particular, some elements such as Mg, Fe3+, and Al3+, as Fe3+/
Fe ratio and grossular molecule demonstrate quite an essential temperature dependence (their correlation coefficients are +0.53, -0.52, and -0.54, and also -0.63, and -0.85 for n=50 at 5% significance level), the others - Ti and
Fe demonstrate from moderate to strong .pressure dependence (for these the correlation coefficients are -0.73, and -0.65, respectively) with a strong positive relationship between themselves (the correlation coefficient 0.90 for n=50). This suggests, in particular, that as the pressure (depth) grows the Fe3+/
Fe ratio has to grow and the concentration of Ti has to decrease in garnets. This mediately follows from the existing schemes of calculation of Fe cations of different valence to the crystallochemical formulae of garnet: Fe3+ =2-(Alvi +Ti) and Fe2+ =Fe - Fe3+ .
The most characteristic regularities revealed in the behaviour of some elements, in particular, Ti, Fe3+/
Fe, and pyrope almandine ratio have been projected onto the supposed stability fields of garnet-bearing mineral associations (see the diagram from the lower continental crust and the upper mantle). The diagram makes it possible to clearly illustrate the specific features of garnet compositions in garnet- bearing rocks of various horizons of the lower continental crust and the upper mantle:
1) garnets characterised by a greater oxidation state of iron ought to occur under greater pressures (at greater depth) as compared with less 'oxidized' garnets. The picture is reversed as to the state of titanation in garnets;
2) garnets formed within continental shield areas and at great depths (Ps>30 kb) ought to be characterised by an increased oxidation of iron and decreased titanium concentration at high pyrope almandine ratio, which is illustrated on the diagram by the intersection of the corresponding isopleth series with the geotherm for the continental regions. Such PT-conditions are not excluded, in particular, for diamond-bearing facies;
3) garnets formed within oceanic blocks and at smaller depths (Ps<30 kb) ought to be characterised by the opposite regularities, which is illustrated by the intersection of the corresponding isopleths with the oceanic geotherm. Such conditions may hold for granulite and eclogite zones of the deep metamorphism.
So, the cited specific features of the behaviour of the isopleths (Fe3+/
Fe, Ti and pyr/alm) in garnets serve as an additional criteria of the estimation and refinement of the PT-conditions of formation of garnet-bearing mineral associations.
References:
90
#
Kotelnikov A.R. , Bychkov A.M. , Chichagov A.V. , Samokhvalova O.L. , and Koval'skii A.M. Synthesis and X-ray study of solid solutions of potassium-rubidium feldspars .key words [potassium rubidium synthesis X-ray study]Institute of Experimental Mineralogy, Russian Academy of Sciences, Chernogolovka, Moscow Region, Russia; Department of Geology, Moscow State University, Moscow, Russia
Solid solutions of potassium-rubidium feldspars were synthesized from gel mixtures under hydrothermal conditions (650-700oC, P = 1-5 kbar). It is shown that mixtures of potassium-rubidium feldspars and leucites are formed at X(Rb) > 0.5, 700oC, and 1 kbar. The temperature decrease to 650oC and the pressure increase to 5.3 kbar enhanced the stability of the feldspars relative to leucite. The conditions and results of experiments on the synthesis of feldspars are presented in Table 1. The X-ray study of (K, Rb)-feldspars was carried out on HZG-4/PC and DRON-1.5 diffractometers. Silicon (spectral purity grade) was used as the internal standard. The unit cell parameters were refined by 30-57 reflections within the angle range from 15 to 85 (2) The parameters were calculated using the LCC and PUDI programs [1] and the MINCRYST information-calculating system of crystal structure data of minerals [2]. The results of the calculations are presented in Table 2. The reflection position of -201 can be used for estimation of the composition of the solid solutions. The accuracy of determination of the composition of (K, Rb)-feldspars is +3 mol.% (Fig. 1).
Table 1. Conditions and results of experiments on the synthesis of (K, Rb)-feldspars from gel mixtures under hydrothermal conditions
|
Entry no |
Gel weighed sample, mg |
(XRb)Fsp (in gel) |
H2O , mg |
ToC |
P, kbar |
Duration, days |
Phase composition after experiment |
|
4513 |
80 |
0.80 |
8 |
680 |
3 |
14.5 |
Fsp+Le |
|
4461 |
524 |
0.70 |
5 |
700 |
1 |
1 |
Fsp+Le |
|
4416 |
2405 |
1.0 |
24 |
700 |
1 |
8 |
Fsp+Le |
|
4463 |
795 |
0.90 |
8 |
700 |
1 |
1 |
Fsp+Le |
|
4464 |
1499 |
1.0 |
15 |
700 |
1 |
1 |
Fsp+Le |
|
4512 |
80 |
0.7 |
8 |
680 |
3 |
14 |
Fsp+Le |
|
4514 |
80 |
0.9 |
8 |
680 |
3 |
14.5 |
Fsp+Le |
|
4515 |
80 |
1.0 |
8 |
680 |
3 |
14 |
Fsp+Le |
|
4552 |
80 |
0.0 |
8 |
680 |
5.2 |
14 |
Fsp |
|
4553 |
80 |
0.2 |
9 |
680 |
5.2 |
14 |
Fsp |
|
4554 |
80 |
0.3 |
8 |
680 |
5 |
14 |
Fsp |
|
4555 |
80 |
0.4 |
9 |
680 |
5 |
14 |
Fsp |
|
4556 |
80 |
0.5 |
10 |
680 |
5 |
14 |
Fsp |
|
4557 |
80 |
0.6 |
9 |
680 |
5 |
14 |
Fsp |
|
4558 |
80 |
0.7 |
9 |
680 |
5 |
14 |
Fsp+Le |
|
4559 |
80 |
0.8 |
10 |
680 |
5 |
14 |
Fsp+Le |
|
4560 |
80 |
0.9 |
6 |
680 |
5 |
14 |
Le |
|
4683 |
225 |
0.5 |
25 |
650 |
5.3 |
17 |
Fsp |
|
4686 |
225 |
1.0 |
25 |
650 |
5.3 |
17 |
Fsp |
# This work was financially supported by the Russian Foundation for Basic Research (Project No. 97-05-65886)
91
Table 2. Unit cell parameters of solid solutions of the sanidine-rubidium sanidine series synthesized under hydrothermal conditions
|
X(1) |
a,� |
b,� |
c,� |
|
V,�3 |
References:* |
|
0.0 |
8.604(1) |
13.030(1) |
7.180(1) |
116.023(4) |
723.3(1) |
1 |
|
0.2 |
8.652(1) |
13.031(1) |
7.182(2) |
116.08(1) |
727.2(1) |
1 |
|
0.3 |
8.674(1) |
13.030(1) |
7.183(1) |
116.09(1) |
729.2(2) |
1 |
|
0.5 |
8.723(2) |
13.034(2) |
7.185(1) |
116.16(2) |
733.3(2) |
1 |
|
1.0 |
8.841(1) |
13.039(2) |
7.194(1) |
116.27(1) |
743.6(2) |
1 |
|
1.0 |
8.841(7) |
13.036(9) |
7.196(4) |
116.26(1) |
744.(1) |
1 |
|
1.0 |
8.837(2) |
13.035(3) |
7.187(1) |
116.270(9) |
742.40(20) |
3 |
|
1.0 |
8.839(1) |
13.034(1) |
7.182(1) |
116.290(9) |
741.80(20) |
4 |
|
0.0 |
8.6045(5) |
13.0306(6) |
7.1787(4) |
116.011(3) |
723.36(7) |
5 |
Note: *(1) X is the molar fraction of the rubidium end member in the solid solution of (K, Rb)-feldspars, References: 1, this work; 3, Voncken et al., 1993; 4, Bruno and Pentinghaus, 1974; 5, Kroll et al., 1986.
References:
Kuz'mina N.A. ,1 Bychkov A.M. ,1 Rusakov V.S. ,2 and Chistyakova N.I. 2 Crystal chemistry, synthesis, and structure transformations of ferrisilicate analogs of feldspars and sparthoids
key words[ferrisilicate feldspar synthesis structure]1 V. I. Vernadskii Institute of Geochemistry and Analytical Chemistry, Russian Academy of Sciences, Moscow, Russia ;2 Department of Physics, Moscow State University, Moscow, Russia
The phases of ferrisilicate analogs of feldspars and feldsparthoids were synthesized from gels under hydrothermal conditions within the 250-475oC temperature range and at pressures from the saturated vapor pressure to 0.5 kbar. The experimental products were studied by the X-ray diffraction and M�ssbauer spectroscopy. The X-ray study included the phase analysis and refinement of the unit cell parameters. The M�ssbauer study included the reduction of the distribution function of hyperfine parameters and simulation processing of the spectrum.
The following important information was obtained on the crystal chemistry of ferrisilicate analogs of feldspars and sparthoids. When the Fe atoms occupy two sites strongly different in local symmetry, the bimodal distribution of hyperfine parameters is observed. In this case, the simulation solution of the spectra makes it possible to estimate the population of each site. This situation was observed for ferrisilicates of the calcilite group in which the difference between quadruple displacements of the partial spectra corresponding to two different sites of iron atoms reached 0.2 mm/s. The unimodal distribution of hyperfine parameters is observed for insignificant differences in local symmetry for tetrahedral sites in ferrisilicates as in ferrisilicate analogs of feldspars and compounds of the leucite group. Such parameters as the distribution width and mean values of hyperfine parameters are the most convenient characteristics for obtaining information on the existence of crystallographically nonequivalent sites. Due to the large ion radius, cesium atoms have the maximum effect on the nearest oxygen environment of the iron atoms, which results in a noticeable increase in the degree of ionic iron-oxygen bond and the corresponding increase in the isomeric shift of the line. This study suggests the long-range effect in the iron and silicon distribution on parameters of hyperfine interactions of the iron nuclei. For example, the isomeric shift for disordered feldspars and compounds of the leucite group is by 0.01 mm/s less than those for ordered feldspar and ordered compounds of the calcilite group regardless of an alkaline cation.
The hydrothermal syntheses of ferrisilicate rubidium leucites and pollucites allowed us to establish that they contain the Fe2+ ions along with the Fe3+ ions. The atomic amount of the Fe2+ ions reach in some samples 14.4% over the spectrum surface area. Therefore, these compounds can be classified as ferrosilicates. The Fe2+ ions appeared in the tetrahedral sites of these silicates due to the reduction of Fe3+ during experiment. The fugacity of oxygen approximately corresponded to that in the buffer Ni/NiO equilibrium. We believe that the necessary condition for the appearance of bivalent iron in the silicate framework, along with the corresponding volatility of oxygen, is tolerance of the phase structure to deviations from stoichiometric ratios, which is typical of leucites, since Fe2+ does not appear under these conditions in the syntheses of feldspars and aegirite.
92
We considered the kinetics of the synthesis and structural transformations for ferrous feldspars. The starting gel is characterized by a substantially wide distribution of the quadruple displacement with a noticeably greater average value of the quadruple displacement. Therefore, the kinetics of crystallization of feldspars was analyzed by a change in average values of hyperfine parameters and the distribution width. The dependences of the synthesis time are qualitatively the same for the both systems. In short experiments, the values of all parameters decrease sharply to a certain value and then monotonically change slightly. The isomeric shifts increase, the widths decrease, and the quadruple shifts remain almost unchanged. The obtained values of rate constants of changing in the average values of the isomeric shifts and quadruple displacements show that the rates of crystallization of potassium feldspar is approximately twofold higher than that of Rb feldspar.
Ordering of the structure of feldspars begins immediately after the formation of their first crystals. The line of the ordering trend for the K phase on going from the monoclinic lattice to the triclinic one has a break, which distinguishes them from alumosilicate feldspars. Only one triclinic sample was obtained for rubidium feldspar, but it also contains, in our opinion, properties of a similar break. This break cannot be explained by simple concentrating of the Fe3+ ions in the corresponding Tl and Tlo sites and by elongation of the Fe-O distance. The reason for the break observed is a change in the joint angles of the T-O-T tetrahedra when the triclinic distortion appears. It is of interest that the unit cell volume of ferrous feldspar increases as the ordering degree increases. The hyperfine parameters of the spectrum changes as the time of the synthesis increases: the isomeric shift increases, and the quadruple displacement and the distribution width decrease for the both (K and Rb) systems. The changes in the hyperfine parameters are low (0.01-0.02 mm/s). The width of the distribution function of the quadruple displacement also decreases by 0.10 mm/s as the time of synthesis increases. Evidently, ordering of Fe and Si in the feldspar framework is the main reason for changes in the hyperfine parameters.
#
Bondar'A.M. ,1 Kozerenko S.V. ,2 and Fadeev2 V.V. Proton magnetic resonance in sulfideskey words[sulphide hydrogen]1 Baikov Institute of Metallurgy, Russian Academy of Sciences, Moscow, Russia ;2 V. I. Vernadskii Institute of Geochemistry and Analytical Chemistry, Russian Academy of Sciences, Moscow, Russia
The presence of hydrogen in the composition of several natural and synthetic iron, molybdenum, zinc, mercury, and other sulfides was established by proton magnetic resonance (1H NMR). The 1H NMR spectra of arsenopyrite recorded by the differential transmission procedure using the technique of nuclear magnetic broad-line resonance are presented in Fig.1. For the majority of samples, the 1H NMR spectra at 24oC exhibited narrow singlet absorption lines with a distance between maxima of the derivative not greater, as a rule, than 0.3-0.5 Gs. At -196oC the lines somewhat broadened but remained singlet. The broad-line 1H NMR data were compared to the results obtained on CXP-180 and FX-100 impulse spectrometers using the high-resolution technique with fast Fourier transformation.
The analysis of the 1H NMR data shows that the signals observed belong most likely to single protons in the crystal structure in the form of groups of the SH-, S2H-, and AsSH2- types; the presence of OH- also cannot be ruled out.
The estimation of the intensity of the signals shows that the content of protons in the sulfides reaches several at.%. The 1H NMR data can explain the observed typical deviations from stoichiometry in sulfides, which are determined by chemical analysis, and suggest the existence of defectness in the anion sublattice of the sulfides (including for the troilite-pyrrhotine series) rather than in the sublattice of iron atoms as has been previously considered.
The presence of protons in the sulfide structures can be a reason for several variations of the semiconductivity parameters of natural sulfides such as photoconductivity, thermal emf, Hall effect, and others. The established presence of protons in the sulfide composition is a rather abundant phenomenon, it can explain several physical and chemical properties of these compounds, such as specific features of phase equilibria, sorption properties, etc., and allows one to consider conditions of formation of sulfide minerals.
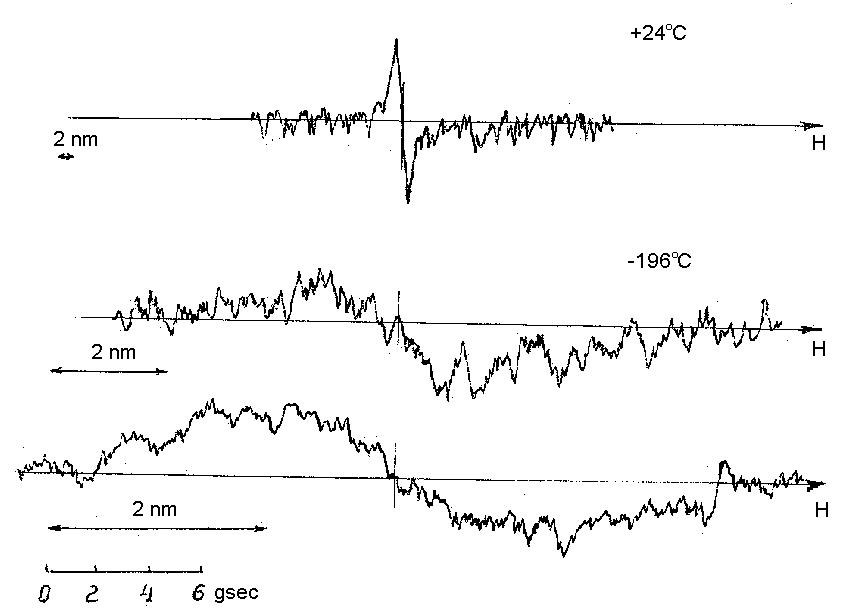
Fig.1. EMR-spectra of arsenopyrite
Ogorodova L.P. , Kiseleva I.A. , Kotelnikov A.R. , and Mel'chakova L.V. Thermodynamic properties of celestine and (Sr,Ba)SO4 solid solution
key words[celestine barite solid solution thermodynamic properties ]Department of Geology, Moscow State University, Moscow, Russia mineral@geol.msu.ru
The variety of barite and celestine compositions in the nature is due to isomorphic Sr-Ba substitutions. Barite
# This work was financially supported by the Russian Foundation for Basic Research (Project No.97-05-64573).
93
with isomorphic substitution of Ba by Sr (up to 8.5%) is abundant, and baritocelestine with the Sr0.56Ba0.37Ca0.06SO4 composition was observed in hydrothermal ores. As a rule, celestine contains small amounts of the Ba component; however, samples with 20% Ba content are also observed. A series of continuous (Sr,Ba)SO4 solid solutions is described in modern deposits of deep ultraacidic sulfate-chloride waters.
The enthalpies of formation of strontium and barium sulfates have not been previously determined by direct calorimetric methods. The values recommended in [1] were calculated from the data on solubility and results of studies of exchange reactions in aqueous solutions at room temperature. The heat capacities of celestine and barite were not measured. Solid solutions of the celestine-barite series were not thermodynamically studied.
Solid samples of celestine, barite, and anhydrite and minerals of solid BaxSr1-xSO4 (x = 0, 0.20, 0.54, 1.0) synthesized under hydrothermal conditions (600, 650oC, 1 kbar) [2] were used for thermochemical studies. The samples were studied by optical, X-ray spectral, and X-ray diffraction methods. The dependences of the unit cell parameters
and
on the composition were established to be linear. The unit cell volumes obey the Wegard rule [2].
The studies were carried out on a Calvert heat conducting high-temperature microcalorimeter (Setaram, France) and a Mettler TA-2000B differential scanning calorimeter (DSC) (Switzerland). The method of dissolvation in the 2PbO*Ba2O3 melt at T = 973 K was used to determine enthalpies of formation and mixing of minerals of solid solutions. The heat contents H0T-H0298.15 of natural celestine in the 590-973 K were measured by the "discharge" method. The heat capacity of natural celestine was measured on DSC in the 110-820 K range in a nitrogen atmosphere with the heating rate of 10 deg/min.
Experiments on dissolvation of the (Sr,Ba)SO4 solid solutions showed that the SrSO4 and Sr0.80Ba0.20SO4 samples were well dissolved, while the BaSO4 and Sr0.46Ba0.54SO4 samples were incompletely dissolved. The enthalpy of dissolvation of barite was estimated from its exchange reaction with carbonates. The negative deviation of the solid solution from ideal ones was observed, which agrees with the results of similar studies of the Sr-Ba isomorphism in the solid solutions of Sr-Ba carbonates [3] and Sr-Ba feldspars [4].
The  T0f,298.15 value for celestine can be calculated from our and corresponding reference data [1] on the exchange reactions:
T0f,298.15 value for celestine can be calculated from our and corresponding reference data [1] on the exchange reactions:
SrSO4 + CaCO3 = CaSO4 + SrCO3 (1)
celestine calcite anhydrite strontianite
SrSO4 + CaO = CaSO4 + SrO (2)
The obtained values of -1457.6 + 4.5 (for reaction (1)) and -1462 + 4.0 (for reaction (2)) kJ/mol agree with each other and data in [1]: -1458.5 + 3.0 kJ/mol obtained previously by other methods. The average of these two results can be recommended for thermodynamic calculations: -1459.8 + 3.0 kJ/mol.
Based on the data on the heat capacity of natural celestine obtained by the DSC method and results of determination of the heat contents on the Calvert microcalorimeter, we calculated the equation of the temperature dependence of the heat capacity of celestine in the 298.15-1000 K range.
References:
94